DOI:
10.1039/C6RA16126J
(Paper)
RSC Adv., 2016,
6, 74757-74764
Best of both worlds: Diels–Alder chemistry towards fabrication of redox-responsive degradable hydrogels for protein release†
Received
21st June 2016
, Accepted 30th July 2016
First published on 3rd August 2016
Abstract
Poly(ethylene glycol)-based redox-responsive hydrogels have been prepared via the Diels–Alder reaction between a furan-containing hydrophilic copolymer and a disulfide-containing bis-maleimide based crosslinker. Hydrogels containing varying amounts of the crosslinker were synthesized with 71–82% yield under mild and benign conditions. Variation in the redox-responsive crosslinker density allows tuning of their mechanical properties and their stability under a reducing environment. It was observed that increase in the amount of crosslinker decreases water uptake and reduces porosity of the hydrogels. Using rheological analysis it was deduced that an increase in temperature and crosslinker density speeds the gelation process and the presence of thiol-containing reducing agents leads to their degradation. Notably, the extent of protein release from these hydrogels could be tailored by varying the amount of redox-responsive disulfide moieties incorporated as crosslinkers during their fabrication.
Introduction
In recent years, hydrogels have emerged as a class of highly desirable crosslinked polymeric materials due to their broad range of applications in areas such as tissue engineering, drug delivery systems, sensors and implant materials.1–5 Increasing interest in hydrogels is due to their simple fabrication, low cost and facile interfacing with various biological materials. The wide utility of these materials necessitates the development of novel design and synthetic approaches towards these materials. Since the advent of ‘click’ chemistry, the design and synthesis, as well as the post-gelation functionalization of hydrogels has undergone noteworthy advancements.6–8 Although initially, the copper catalyzed Huisgen-type azide–alkyne cycloaddition was widely utilized for facile and efficient synthesis of hydrogels,9,10 recent focus involves the use of metal-free conjugation based strategies. The strain promoted azide–alkyne cycloaddition,11 nucleophilic thiol–ene,12 radical thiol–ene13 and the Diels–Alder reactions14 are the most commonly utilized metal-free reactions that have been used to obtain hydrogels. Among the various metal-free ‘click’ chemistries, the Diels–Alder (DA) cycloaddition provides a robust conjugation methodology that does not require utilization of any catalyst or photochemical irradiation, and generally involves diene and dienophiles as functional groups that are quite stable and non-reactive, when compared to reactive functional moieties such as azides and thiols. Because of these attractive characteristics, the DA cycloaddition has been employed in fabrication of various polymeric materials.15–17 Furthermore, the DA reaction is also thermoreversible, hence, has been widely used to obtain self-healing materials.18–23 Over past decades, several studies have used polymers containing furan moieties as side chains to fabricate dynamic and self-healing materials. Recently, Sumerlin and coworkers exploited the dynamic nature of this cycloaddition to design a system that allows switching between linear polymers and branched multi-arm macromolecular constructs.24 Furan appended linear diblock copolymers upon reaction with a bis-maleimide based crosslinker assembles to a multi-arm star copolymer, which upon heating reverts back to their linear form. In another example, Singha and coworkers demonstrated that copolymers containing pendant furan groups can be crosslinked using bismaleimide linkers to obtain films that repair upon heating when scratched.25,26 Recent years have also witnessed an ever increasing utilization of the DA reaction as a ‘click’ reaction of choice for synthesis of hydrogel.27–29 Recently, Goepferich and coworkers made vital contributions towards fabrication of hydrogels using the DA chemistry between furan and maleimide functionalized multi-arm PEG polymers.30–32 Utilization of the DA chemistry as a choice for fabrication of hydrogels is particularly attractive since the DA reaction between diene and dienophile is accelerated in aqueous media.33–35 Although the DA reaction is attractive for hydrogel formation, a particular limitation stems from the fact that degradability of such hydrogels can be achieved at relatively high temperatures around 100 °C that is required for the retro-Diels–Alder reaction.
To date, several stimuli-responsive triggers such as UV-irradiation, enzymes, pH, temperature, redox potential and ultrasound have been incorporated into various polymeric materials to facilitate their degradation at physiological conditions.36–42 Among these, redox responsive materials based on disulfide linkages have drawn a lot of attention over past years because of their enhanced degradation due to the increased amount of glutathione present in the vicinity of tumors.43,44 A whole range of polymeric drug delivery systems such as micelles,45–48 nanogels49,50 and network structures51 reported in recent literature incorporates the thiol-responsive disulfide trigger. Recently, Grayson and coworkers reported poly(hydroxyethyl methacrylate) based hydrogels containing disulfide linkages that showed substantial increased sustained release rates for encapsulated dyes when compared to release from hydrogels fabricated using non-responsive crosslinkers.52 Another example of redox-responsive hydrogels was reported by Du Prez and coworkers who designed PEG-based cryogels containing degradable disulfide linkages that underwent efficient degradation in glutathione containing media.53
Given the positive attributes of using the metal-free Diels–Alder cycloaddition reaction to fabricate hydrogels and the disulfide exchange reaction as a handle to induce degradation, we designed a hydrogel system that benefits from both of these reactions. Herein, we outline a strategy to fabricate degradable hydrogels for tunable protein release, using the Diels–Alder chemistry for fabrication while incorporating a disulfide–thiol exchange chemistry based redox responsive degradation trigger (Scheme 1). The synthesis of hydrogels is carried out at physiological temperature in aqueous media under benign conditions. It can be expected that the variation in the redox-responsive crosslinker density should allow tuning of mechanical properties and stability under reducing environment, as well as modulation of protein release from these hydrogels.
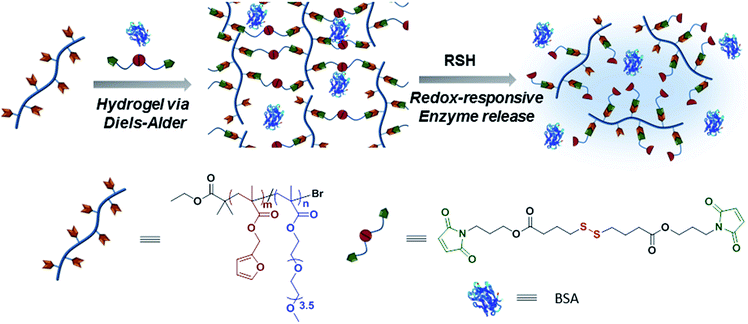 |
| | Scheme 1 Schematic illustration of synthesis and degradation of hydrogels. | |
Experimental
Materials
Maleic anhydride was purchased from Merck. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) (98+%), 1,4-dithio-DL-threitol (DTT) (98%), copper(II) bromide (98%) were purchased from Alfa Aesar. 4-(Dimethylamino)pyridine (>99%), copper purum p.a. (>99%), furan (>99%), 3-amino-1-propanol (>99%), 4,4′-dithiodibutyric acid (95%), furfuryl methacrylate (97%), poly(ethylene glycol)methyl ether methacrylate (PEGMEMA, Mn 300), ethyl α-bromoisobutyrate (98%), tris[2-(dimethylamino)ethyl]amine (Me6TREN) (97%) N,N-dimethylformamide (DMF) (99.8%) were purchase from Sigma-Aldrich. The furan–maleic anhydride adduct and furan-protected maleimide-containing alcohol were prepared according to literature procedures.54
Measurement
Crosslinker characterization was carried out with 1H NMR spectroscopy (Varian 400 MHz). The molecular weight of the copolymers were determined by size exclusion chromatography using Shimadzu PSS-SDV (length/ID 8 × 300 mm, 10 μm particle size) linear Mixed C column calibrated with polystyrene standards using a refractive-index detector. Tetrahydrofuran (THF) was used as eluent at a flow rate of 1 mL min−1 at 30 °C. Microstructures of the hydrogels were investigated with JEOL NeoScope JCM-5000 scanning electron microscopy (SEM) instrument at an accelerating voltage of 10 kV. Hydrogel rheological behaviors were evaluated by measuring the loss (GII) and storage (GI) moduli of the hydrogel as a function of angular frequency and time using an Anton PAAR MCR 302 rheometer. Fluorescein isothiocyanate (FITC)-labeled bovine serum albumin (BSA) release from hydrogels was analyzed using Varian Cary 50 Scan UV/vis spectrophotometer.
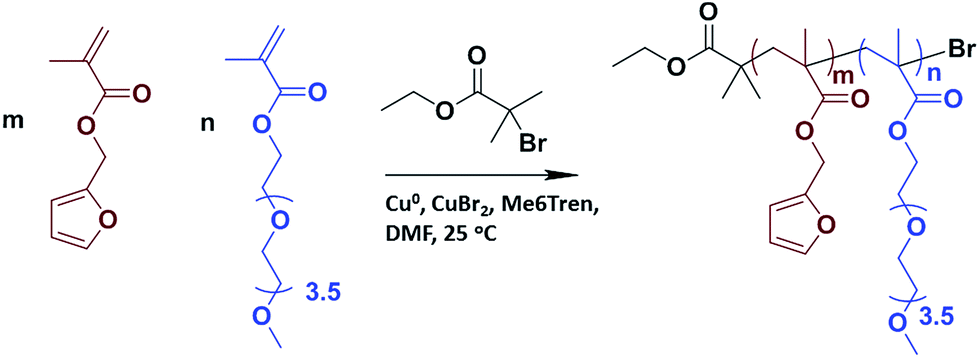
Synthesis of furan-containing copolymer
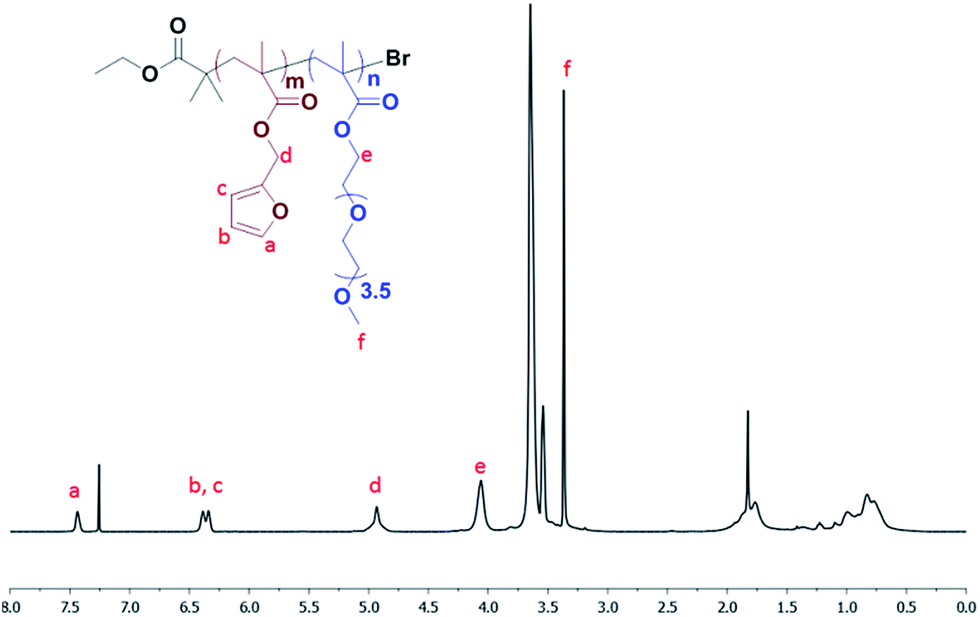
The ratio of reactants used were [monomers]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[initiator]:[CuBr2]:[Me6TREN]: 10:1:0.05:0.12 (molar equivalents). Furfuryl methacrylate (97% purity, 0.2 mL, 1.3 mmol), PEGMEMA (average Mn 300, 0.8 mL, 2.8 mmol), ethyl α-bromoisobutyrate (98% purity, 0.048 mL, 0.3 mmol) and solvent (DMF, 0.6 mL) were added to a round bottom flask. Copper(II) bromide (98% purity, 0.004 g, 0.002 mmol), Me6TREN (97% purity, 0.009 g, 0.004 mmol) and solvent (DMF, 0.5 mL) were added to a different round bottom flask. Both solutions were purged with N2 for 30 minutes. After 30 minutes, solutions were mixed and polymerization was started. To remove the copper from the product solution, the resulting mixture was passed over a basic alumina column, with dichloromethane as eluent. The product was concentrated on a rotary evaporator and precipitated in cold ether. 1H NMR (CDCl3, δ, ppm), 7.44 (s, 1H, CH
:[initiator]:[CuBr2]:[Me6TREN]: 10:1:0.05:0.12 (molar equivalents). Furfuryl methacrylate (97% purity, 0.2 mL, 1.3 mmol), PEGMEMA (average Mn 300, 0.8 mL, 2.8 mmol), ethyl α-bromoisobutyrate (98% purity, 0.048 mL, 0.3 mmol) and solvent (DMF, 0.6 mL) were added to a round bottom flask. Copper(II) bromide (98% purity, 0.004 g, 0.002 mmol), Me6TREN (97% purity, 0.009 g, 0.004 mmol) and solvent (DMF, 0.5 mL) were added to a different round bottom flask. Both solutions were purged with N2 for 30 minutes. After 30 minutes, solutions were mixed and polymerization was started. To remove the copper from the product solution, the resulting mixture was passed over a basic alumina column, with dichloromethane as eluent. The product was concentrated on a rotary evaporator and precipitated in cold ether. 1H NMR (CDCl3, δ, ppm), 7.44 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHO), 6.39 (d, 2H, furan H's), 4.93 (s, 2H, COOCH2C), 4.06 (s, 2H, COOCH2CH2O), 3.37 (s, 3H, OCH3).
CHO), 6.39 (d, 2H, furan H's), 4.93 (s, 2H, COOCH2C), 4.06 (s, 2H, COOCH2CH2O), 3.37 (s, 3H, OCH3).
Synthesis of maleimide-based disulfide-containing crosslinker
Furan-protected maleimide-containing alcohol (4.96 g, 22 mmol),54 4,4′-dithiodibutyric acid (1.14 g, 5.5 mmol), EDCI (3.37 g, 17.6 mmol) and 4-dimethylaminopyridine (DMAP) (0.403 g, 4.65 mmol) were dissolved in dry dichloromethane (15 mL). The solution was stirred for 24 hours at room temperature. Dichloromethane (25 mL) added to reaction mixture and organic phase was washed with saturated NaHCO3. The combined organic layers were dried over anhydrous Na2CO3 and concentrated solution was purified by column chromatography on SiO2 (EtOAc:CH2Cl2 1:2) affording 2.73 g (80% yield) of the furan protected bismaleimide crosslinker. 1NMR (CDCl3, δ, ppm) 7.24 (s, 4H, CHCH), 5.19 (s, 4H, CH bridgehead protons), 3.98 (t, 4H, OCH2CH2), 3.51 (t, 4H, NCH2CH2), 2.77 (s, 2H bridge protons), 2.67 (t, 4H, SCH2CH2), 2.39 (t, 4H, COCH2CH2), 1.97 (m, 4H, SCH2CH2CH2), 1.85 (m, 4H, NCH2CH2CH2). Retro Diels–Alder reaction was carried out by dissolving compound furan protected bismaleimide crosslinker (2.73 g) in toluene (10 mL). The solution is heated to 110 °C to reflux 24 hours. The concentrated solution was purified by flash chromatography on SiO2 (EtOAc:CH2Cl2 1:1). 1.94 g bismaleimide crosslinker is obtained with 90% yield. 1NMR (CDCl3, δ, ppm), 6.7 (s, 4H, CHCH), 4.07 (t, 4H, OCH2CH2), 3.63 (t, 4H, NCH2CH2), 2.73 (t, 4H, SCH2CH2), 2.44 (t, 4H, COCH2CH2), 2.03 (m, 4H, SCH2CH2CH2), 1.95 (m, 4H, NCH2CH2CH2); 13C NMR (CDCl3) 173.1, 170.5, 134.6, 61.5, 38.6, 35.5, 33.7, 27.8, 24.5.
Preparation of hydrogels
Hydrogels were prepared with three different crosslinker concentrations. Copolymer (100 mg) and different amount of crosslinker (10/4, 10/6, 10/8, furan/maleimide molar ratio) were dissolved in dioxane:water (1:4 v/v, 1 mg μL−1). The mixture was sonicated for 2 minutes before placing the vial in an oil bath at preset temperature. For loading of FITC-BSA, the hydrogel precursors were added into a protein containing dioxane/water solution. The solution was sonicated for 10 minutes for homogenous mixing before gelation at 37 °C. The hydrogel samples were rinsed with water before release studies to remove any adhered biomolecules.
Characterization of hydrogels
Gelation yields of hydrogels. Gelation yield of hydrogels were calculated gravimetrically. According to time sweep test in rheometer, end point of gelation process was determined. The point which elastic modulus reaches the highest point was accepted as a highest conversion of the process. After this point, hydrogels were first washed with excess amount of tetrahydrofuran then water to remove unreacted polymer and crosslinker. Afterwards, water was removed with lyophilizer. Dried hydrogels masses (Mdried) was used to calculate yield of gelation processes as (Mdried/Mp+c) × 100 where Mp+c was the total mass of polymer (p) and crosslinker (c).
Swelling of hydrogels. Lyophilized hydrogel disks (80 mg) were immersed in deionized water at room temperature. The hydrogel was weighed after removal of water on the surface at periodic time points. Water uptake experiment was continued until no further increase in hydrogel weight was observed. Degree of swelling were calculated from three different experiment for each hydrogels as (Ms/Mdry) × 100 where Ms and Mdry refers to weight of swollen and dry hydrogels, respectively.
Rheological analysis of hydrogels. The effect of crosslinker density and temperature on physical properties of hydrogels was investigated with Anton Paar MCR 302 rheometer. The test geometry was 15 mm diameter plate. Strain sweep test was applied to solid state hydrogel to check the linear-viscoelastic limit regime (LVR). Nonlinear strain regions represent the structural breakdown in hydrogels which were out of LVR range.55 Frequency sweep test allows us to identify the lower frequency limit that solid state hydrogel behavior can be observed. Frequency sweep test with an appropriate strain value allowed choice of a frequency range to undertake time sweep tests. Gelation process was followed by time sweep test with testing parameters as strain value of 0.5% and angular frequency value of 10 rad s−1. Time sweep test was run by filling gap between preheated rheometer plates with liquid state polymer–crosslinker mixture. To prevent solvent escape, a closed system was used for gelation studies.
Redox-responsive degradation of hydrogels
Disk shaped hydrogels (2 mm thick, 12 mm diameter, 80 mg) were incubated at 37 °C in a thermal shaker with 200 rpm in aqueous medium (PBS, 3 mL) containing 21 × 10−3 M DTT. After 24 hours, solution was changed with a fresh DTT containing PBS solution. A digital camera was used to record the degradation process of bulk hydrogels.
Protein release in vitro
Fluorescein isothiocyanate (FITC)-labeled bovine serum albumin (BSA) was encapsulated into hydrogels as described above. After gelation, surface of hydrogels were washed with copious amounts of water. BSA containing hydrogel disks (2 mm thick) were divided into 30 mg pieces and placed into PBS (2 mL) and PBS containing DTT (21 mM, 2 mL) at 37 °C. Solutions were periodically replaced with fresh solutions and the amount of protein release in the supernatant was determined using UV-vis spectrophotometer. Three different release experiments were performed for each hydrogels.
Results and discussion
Synthesis and characterization of hydrogel precursors
Synthesis of redox-sensitive bismaleimide-containing crosslinker. Disulfide containing bismaleimide based crosslinker was synthesized through esterification of 4,4′-dithiodibutyric acid with a furan-protected maleimide-containing alcohol in the presence of EDCI and DMAP (Fig. S2†). The maleimide groups were unmasked by removal of the furan moiety at 110 °C via the retro Diels–Alder reaction. The composition and purity of the crosslinker was deduced from the 1H NMR spectrum where the presence of proton resonances at 6.7 ppm and 2.75 ppm belonging to vinylic proton on the maleimide ring and protons adjacent to the disulfide unit, respectively, confirms the structure (Fig. S3†). Additionally, 13C NMR analysis was also used to validate the structure of thus obtained crosslinker (Fig. S4†).
Preparation and characterization of hydrogels
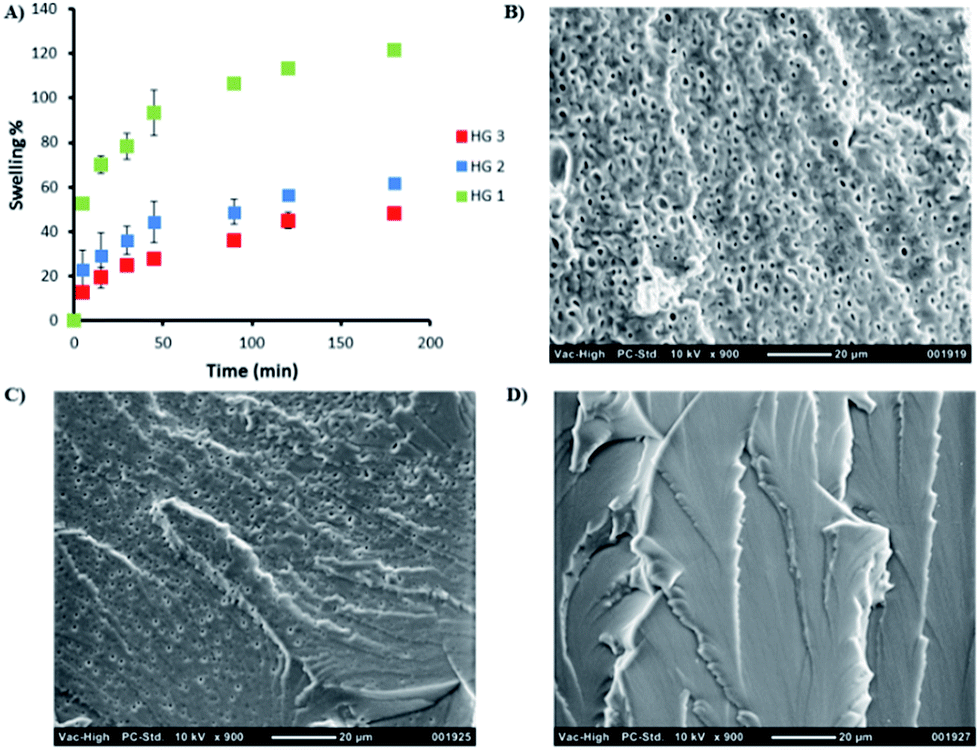
A series of hydrogels (HG1–HG3) with varying amounts of crosslinking were synthesized by adjusting the molar ratio of maleimide units from the crosslinker to number of furan groups on the polymeric chain (Table 1). Clear and transparent hydrogels were prepared by mixing the polymer and crosslinker in a water/dioxane (4:1) mixture at 37 °C (Scheme 2). Dioxane was added as a co-solvent in order to facilitate the solubility of the bis-maleimide crosslinker as well as it was observed that it also remarkably reduces the hydrolysis of the maleimide group. Stability of the bismaleimide crosslinker in PBS and 20% PBS–dioxane mixture was analyzed using UV-vis spectroscopy to reveal that while the linker is stable in PBS–dioxane mixture, it slowly undergoes hydrolysis in PBS solution during gelation time (Fig. S5†). The hydrogel compositions and conversions of gelation are summarized in Table 1. Obtained results suggest a slight increase in conversions with an increment in crosslinker. Furthermore, hydrogels with different crosslinker density can be expected to possess different physical properties such as extent of swelling, stiffness and stability. Swelling degree of hydrogels can be adjusted by changing the amount of crosslinker during the gelation. Comparison of swelling profiles of these hydrogels in water shows that a decrease in the crosslinker density dramatically increases their swelling (Fig. 3A). Likewise, investigation of morphology of these hydrogels using SEM suggests that their porosity is also dependent on the extent of crosslinking. For example, hydrogel HG1 fabricated with less crosslinker clearly possesses a more porous structure compared to the hydrogel HG3 that exhibits an almost nonporous structure (Fig. 3). Rheological investigations were undertaken to probe the variation in stiffness of hydrogels by changing the extent of crosslinking. In this regard, hydrogel samples were subjected to strain sweep test, frequency sweep test and time sweep test. Strain sweep test is recommended for obtaining the linear viscoelastic limits of hydrogel.55 When 0.01 to 100% strain was applied on hydrogel HG2, a linear behavior for all strain values was observed which indicates lack of structural breakdown for all strain values (Fig. S6†). Stiffness behavior of hydrogel was measured using frequency sweep test from 0.01 to 100 Hz by using strain value of 0.5 (Fig. 4A). All hydrogels showed expected gel behavior and storage modulus of hydrogels increased with crosslinking. Time sweep tests were performed by using strain of 0.5 and frequency of 10 rad s−1 to follow the gelation process, estimate the gelation time and final elastic modulus for hydrogels with varying crosslinker density. Initially, the storage modulus could not be observed because it was very low; hence, it was only just before the onset of gelation it was measurable with the instrument during the time sweep test. Gelation point is indicated by the crossover of storage modulus and loss modulus. First, the gelation processes of hydrogel HG3 was studied at 25 °C, 30 °C and 37 °C. It was observed that the onset of gelation occurred earlier at 37 °C than at 25 °C, presumably due to the increased rate of Diels–Alder reaction at higher temperatures (Fig. 4C). Time sweep tests were also used to investigate the effect of amount of crosslinker on gelation point and storage modulus. It was observed that the onset of gelation occurs earlier upon increasing the amount of crosslinker (Fig. 4B).
Table 1 Hydrogels fabricated with varying amounts of crosslinker
| Hydrogel |
[M]1/[M]2a |
Conversionb |
| Theoretical mol equivalent; [M]1: furan, [M]2: maleimide. Measured gravimetrically. |
| HG1 |
10/4 |
72 |
| HG2 |
10/6 |
77 |
| HG3 |
10/8 |
81 |
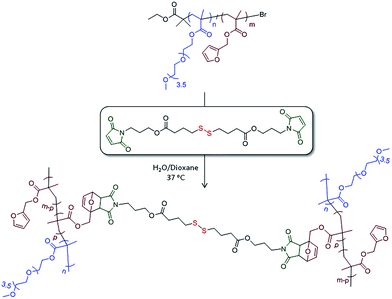 |
| | Scheme 2 Illustration of Diels–Alder cycloaddition mediated gelation. | |
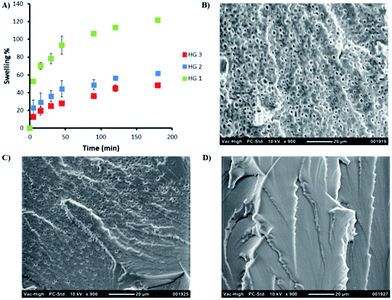 |
| | Fig. 3 (A) Effect of crosslinker density on the degree of swelling of hydrogels (B) SEM micrograph of HG1, (C) SEM micrograph of HG2, and (D) SEM micrograph of HG3. | |
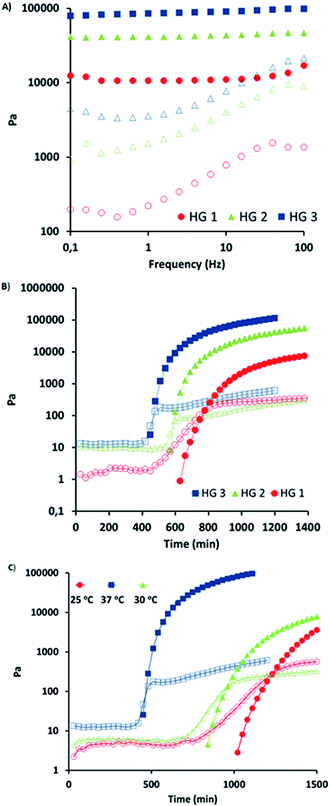 |
| | Fig. 4 (A) Frequency sweep test for hydrogels (HG1–HG3) at 37 °C, (B) time sweep test for hydrogels with different crosslinker density at 37 °C, and (C) time sweep test of HG3 at different temperatures (filled markers: GI, empty markers: GII). | |
Degradation of redox-responsive hydrogels
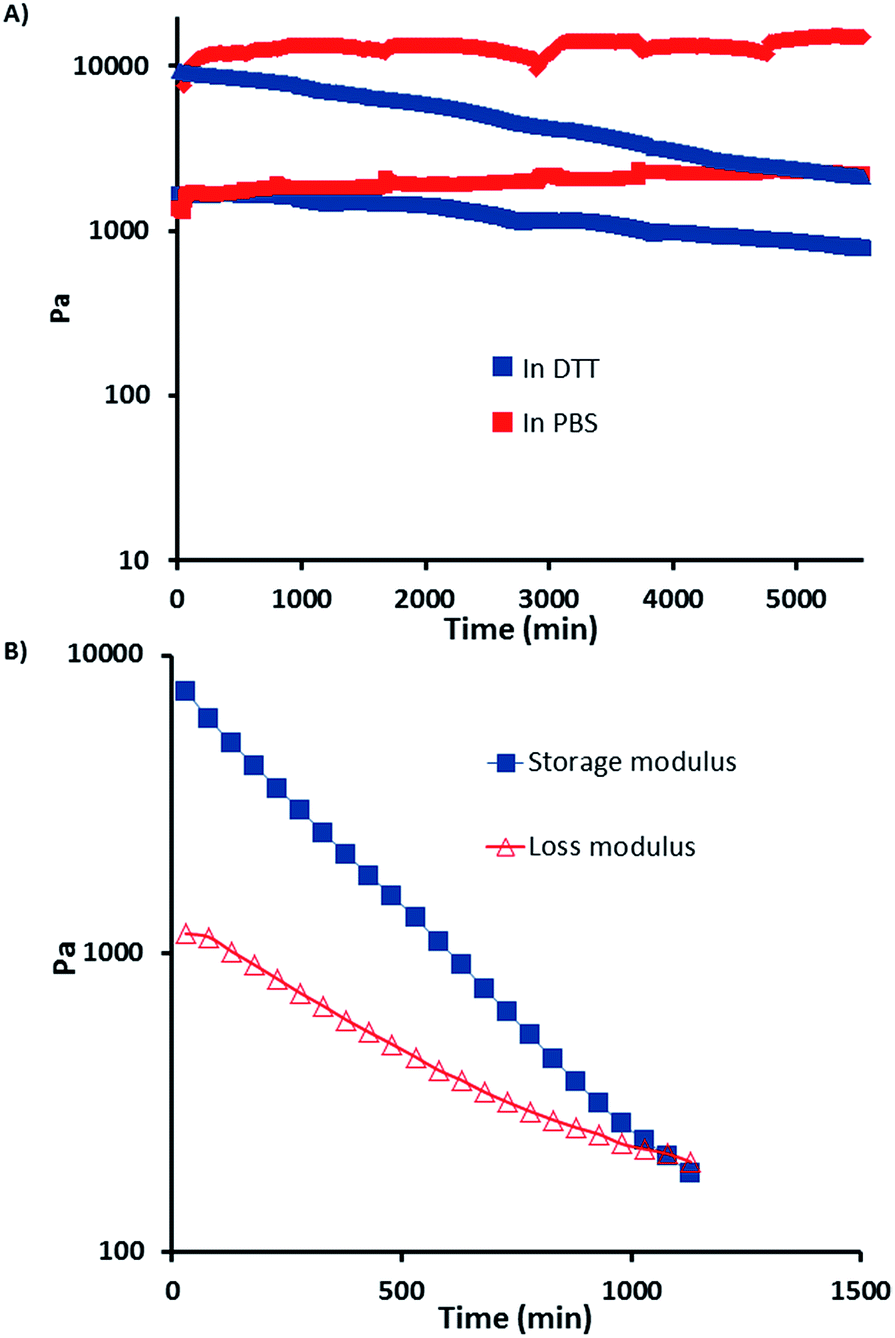
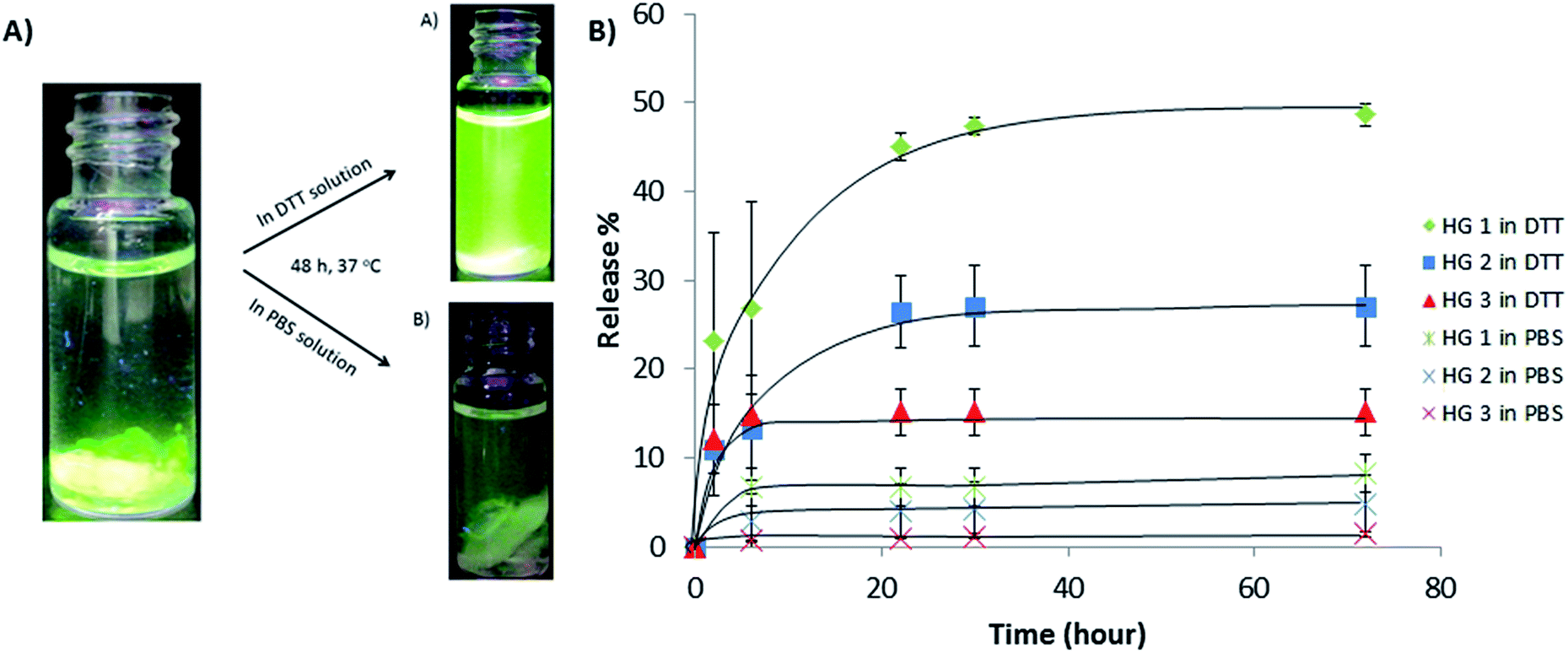
These hydrogels contain redox responsive disulfide bonds and reversible Diels–Alder endo product at the same time. Although there may be some cycloadducts with endo stereochemistry that has a relatively low cycloreversion temperature, these hydrogels were found to be quite stable under non-reducing ambient conditions. It is well established that disulfide bonds can be degraded in the presence of reducing agents such as DTT. In order to probe their stability and thiol-responsive degradation, disk shaped dry hydrogel samples of HG1 were immersed in a PBS solution and a DTT containing (21 mM) PBS solution at 37 °C. It was observed that while the hydrogel immersed in DTT solution lost its shape and became very soft due to reduction in crosslinking, the hydrogel immersed in PBS solution did not undergo appreciable degradation due to preservation of disulfide linkages in the absence of reducing agent (Fig. 5). Degradation of hydrogel under reducing condition was also followed with time sweep test (Fig. 6A). These experiments were done by immersing hydrogels in PBS and DTT (21 mM and 200 mM) containing PBS solutions. Loss in storage modulus of the hydrogel imbibed with 21 mM DTT solution suggested that hydrogels lose their stiffness due to reduction in crosslinking. Additionally, when the concentration of DTT is increased nearly 10-fold (to 200 mM), the storage modulus (GI) gradually decreases and a cross-over point was observed between the loss modulus (GII) and GI (Fig. 6B), where the hydrogel loses its gel like structure and exhibits sol-like behavior. Increased concentration of DTT (200 mM) was used for this experiment to observe this crossover point in a reasonable time period using rheology.
 |
| | Fig. 5 Degradation of disulfide-containing hydrogel (HG1) in PBS and DTT. | |
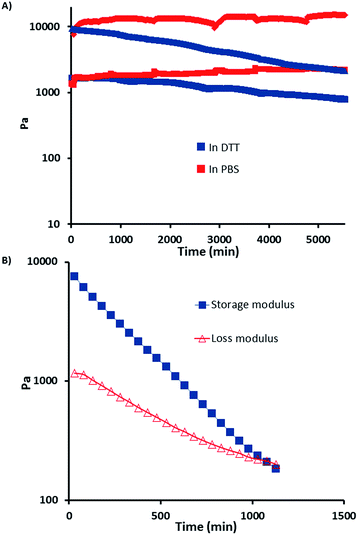 |
| | Fig. 6 (A) Change in storage modulus for hydrogel (HG1) at 37 °C in PBS and DTT (21 mM) solution, (red markers: GI, blue markers: GII), undulations are due to solvent replenishments. (B) Rheological investigation of gel- to sol-form for hydrogel (HG1) due to disulfide bond cleavage observed in the presence of 200 mM DTT. | |
Behavior of hydrogels in the PBS solution devoid of any DTT can be acceptable as a control experiment. Fig. 6A shows that hydrogel HG1 maintains its mechanical stability in PBS solution.
As a next step, we probed if we can use variation in crosslinker to control the release of biomolecules encapsulated within hydrogels.
Encapsulation and release of protein in redox-responsive hydrogels
Hydrogels varying in the amount of degradable crosslinker were utilized to study protein release using FITC-labeled BSA. The protein was encapsulated within the hydrogel matrix during their formation. After gelation, hydrogels were washed with water and non-encapsulated BSA in the aqueous media was determined using UV-vis spectroscopy to calculate the amount of protein trapped within the hydrogels. Next, we investigated the release of FITC-BSA from these hydrogel by immersing them in PBS solutions in the presence and absence of DTT. Higher release of FITC-BSA molecules into the aqueous media in the presence of DTT compared was obvious due to its increased fluorescence, when the vials were irradiated with UV light (Fig. 7A). To ascertain the effect of amount of crosslinker, hydrogels (HG1, HG2, and HG3) were immersed in PBS and DTT containing PBS solution (21 mM). Periodically, aliquots were collected for measurement of released BSA using UV-vis spectroscopy. It was observed that the cumulative release of BSA was remarkably higher than release in PBS alone for all hydrogels (Fig. 7B). Furthermore, comparison of the BSA release profiles from the different hydrogels clearly highlights the role played by the cleavable linker. As expected, less release was observed for the more crosslinked and stiffer hydrogel HG3, while maximum BSA release was observed for hydrogel HG1 containing least amount of the crosslinker.
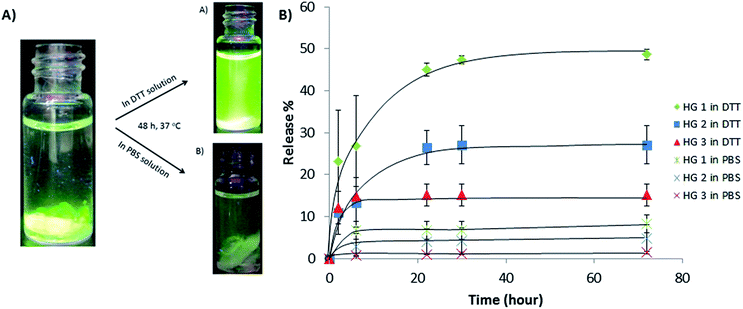 |
| | Fig. 7 (A) Visual photographs of UV-illuminated vials demonstrating differences in the release of FITC-BSA from hydrogels in DTT (21 mM) and in PBS solutions. (B) Release profile of FITC-BSA from hydrogels with varying crosslinking in DTT and PBS solution at 37 °C. | |
Conclusions
Redox-responsive hydrogels were synthesized using furan-containing hydrophilic PEG-based copolymers and a disulfide-containing maleimide based crosslinker. The furan-containing copolymer was obtained through copolymerization of FuMA and PEGMEMA using SET-LRP. The novel disulfide containing bis-maleimide crosslinker installs the redox-responsive linkages during gelation through the Diels–Alder reaction in aqueous media. Hydrogels with varying extent of crosslinking were fabricated and gelation process was followed using their rheological analysis. Degradation studies demonstrated that thus fabricated hydrogels readily degraded in a solution containing DTT, a reducing agent, and this was also confirmed by rheological methods. Finally, it was observed that control over crosslink density within the hydrogels using the redox-responsive linker enables modulation of protein release profiles.
Acknowledgements
Authors thank The Ministry of Development of Turkey (Project 2009K120520) for their contribution to the infrastructure.
Notes and references
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- B. D. Ratner and A. S. Hoffman, Hydrogels for Medical and Related Applications, 1976, vol. 31 Search PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- S. Ahadian, R. B. Sadeghian, S. Salehi, S. Ostrovidov, H. Bae, M. Ramalingam and A. Khademhosseini, Bioconjugate Chem., 2015, 26, 1984–2001 CrossRef CAS PubMed.
- D. Caccavo, S. Cascone, G. Lamberti and A. A. Barba, Mol. Pharm., 2015, 12, 474–483 CrossRef CAS PubMed.
- S. Yigit, R. Sanyal and A. Sanyal, Chem.–Asian J., 2011, 6, 2648–2659 CrossRef CAS PubMed.
- C. M. Nimmo and M. S. Shoichet, Bioconjugate Chem., 2011, 22, 2199–2209 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- M. Clark and P. Kiser, Polym. Int., 2009, 58, 1190–1195 CrossRef CAS.
- N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS PubMed.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4103–4120 CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- A. P. Bapat, J. G. Ray, D. A. Savin, E. A. Hoff, D. L. Patton and B. S. Sumerlin, Polym. Chem., 2012, 3, 3112–3120 RSC.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 CAS.
- N. B. Pramanik, D. S. Bag, S. Alam, G. B. Nando and N. K. Singha, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3365–3374 CrossRef CAS.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- H. Tan, J. P. Rubin and K. G. Marra, Macromol. Rapid Commun., 2011, 32, 905–911 CrossRef CAS PubMed.
- F. Yu, X. Cao, L. Zeng, Q. Zhang and X. Chen, Carbohydr. Polym., 2013, 97, 188–195 CrossRef CAS PubMed.
- S. Kirchhof, A. Strasser, H.-J. Wittmann, V. Messmann, N. Hammer, A. M. Goepferich and F. P. Brandl, J. Mater. Chem. B, 2014, 3, 449–457 RSC.
- S. Kirchhof, F. P. Brandl, N. Hammer and A. M. Goepferich, J. Mater. Chem. B, 2013, 1, 4855–4864 RSC.
- N. Hammer, F. P. Brandl, S. Kirchhof, V. Messmann and A. M. Goepferich, Macromol. Biosci., 2014, 3, 405–413 Search PubMed.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- S. Otto and J. B. Engberts, Org. Biomol. Chem., 2003, 1, 2809–2820 CAS.
- H.-L. Wei, Z. Yang, L.-M. Zheng and Y.-M. Shen, Polymer, 2009, 50, 2836–2840 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- S. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Polym. Chem., 2015, 6, 5565–5574 RSC.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- K. C. Koehler, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- Z. A. A. Hamid, A. Blencowe, B. Ozcelik, J. A. Palmer, G. W. Stevens, K. M. Abberton, W. A. Morrison, A. J. Penington and G. G. Qiao, Biomaterials, 2010, 31, 6454–6467 CrossRef CAS PubMed.
- N. R. Ko and J. K. Oh, Biomacromolecules, 2014, 15, 3180–3189 CrossRef CAS PubMed.
- A. Cunningham, N. R. Ko and J. K. Oh, Colloids Surf., B, 2014, 122, 693–700 CrossRef CAS PubMed.
- N. Chan, N. Yee, S. Y. An and J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2057–2067 CrossRef CAS.
- N. Chan, S. Y. An and J. K. Oh, Polym. Chem., 2014, 5, 1637–1649 RSC.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- J. Groll, S. Singh, K. Albrecht and M. Moeller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5543–5549 CrossRef CAS.
- B. Gyarmati, Á. Némethy and A. Szilágyi, Eur. Polym. J., 2013, 49, 1268–1286 CrossRef CAS.
- M. Ejaz, H. Yu, Y. Yan, D. A. Blake, R. S. Ayyala and S. M. Grayson, Polymer, 2011, 52, 5262–5270 CrossRef CAS.
- T. Dispinar, W. Van Camp, L. J. De Cock, B. G. De Geest and F. E. Du Prez, Macromol. Biosci., 2012, 12, 383–394 CrossRef CAS PubMed.
- B. J. Neubert and B. B. Snider, Org. Lett., 2003, 5, 765–768 CrossRef CAS PubMed.
- J. M. Zuidema, C. J. Rivet, R. J. Gilbert and F. A. Morrison, J. Biomed. Mater. Res., Part B, 2014, 102, 1063–1073 CrossRef PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4917–4926 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16126j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.