
Study on the oxidation process of cobalt hydroxide to cobalt oxides at low temperatures
Abstract
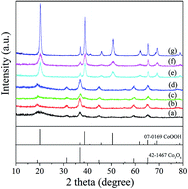
The transformation process of cobalt hydroxide (Co(OH)2) to cobalt oxides (Co3O4/CoOOH) in aqueous solution was studied in the temperature range of 50–90 °C. The crystalline structures and morphologies of the samples were characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR) and scanning electron microscopy (SEM) analysis. The OH− : Co2+ ratio and aging time influenced the transformation process from Co(OH)2 to final products. And the formation of hydrotalcite-like phase intermediate ([CoII1−xCoIIIx(OH)2](NO3)x·nH2O) is a crucial factor for synthesizing Co3O4. When the OH− : Co2+ ratio was in the range of 1.0 : 1–1.8 : 1, α-Co(OH)2 was oxidized to [CoII1−xCoIIIx(OH)2](NO3)x·nH2O at first, and then transformed to Co3O4 crystals. When the OH− : Co2+ ratio was in the range of 2.0 : 1–50.0 : 1, without the appearance of soluble Co(OH)42− ions in the reaction system before oxidation, fast conversion from α-Co(OH)2 to β-Co(OH)2 resulted in the formation of only CoOOH crystals, without the appearance of [CoII1−xCoIIIx(OH)2](NO3)x·nH2O or Co3O4 crystals.
 Please wait while we load your content...
Please wait while we load your content...

