
Highly mesoporous CsTaWO6via hard-templating for photocatalytic hydrogen production†
Abstract
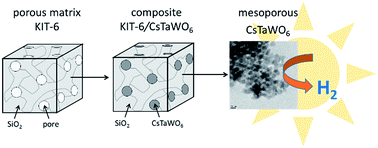
The quaternary photocatalyst CsTaWO6 was for the first time prepared with a high surface area of 115 m2 g−1 via a hard-templating approach. The highly crystalline and phase-pure material was applied in photocatalytic hydrogen production experiments to demonstrate the influence of porosity, surface area and crystallinity on charge carrier transfer.
 Please wait while we load your content...
Please wait while we load your content...

