
ZnO rods rooted on manifold carbon nanofiber paper as a scalable photocatalyst platform: the effects of ZnO morphology†
Abstract
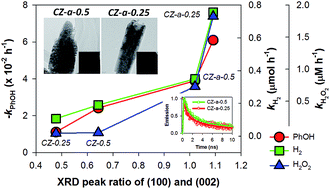
Crystalline ZnO rods rooted on manifold carbon nanofiber (CNF) paper were synthesized via electrodeposition of ZnO onto electrospun CNF paper (∼300 μm thick) followed by oxidative annealing. The morphology of the ZnO deposited on the conductive CNF paper can be tailored to be polycrystalline rods with convex-shaped ends at a high Zn2+ precursor concentration (0.5 mM) upon annealing (denoted CZ-a-0.5, where “a” refers to annealing), whereas the sample electrodeposited at a low Zn2+ precursor concentration (0.25 mM) results in single crystalline rods with concave-shaped ends (denoted CZ-a-0.25). In order to systematically examine the photocatalytic activity, the annealed and non-annealed CNF/ZnO samples (CZ-a and CZ, respectively) were compared for the oxidation of phenol (one-electron transfer reaction), the production of H2 via water splitting and H2O2 production via oxygen reduction (both two-electron transfer reactions). Irrespective of the type of reaction, the CZ-a samples exhibit superior photocatalytic activities than the CZ samples in the following order: CZ-a-0.25 > CZ-a-0.5 > CZ-0.5 > CZ-0.25. The observed activity order is consistent with the trend observed in the XRD intensity ratio between the (100) and (002) planes (i.e., I100/I002 ratio) of the corresponding samples. The time-resolved photoluminescence decay spectra further reveal that the average lifetime of charge carriers is the shortest for CZ-a-0.25, followed by CZ-a-0.5, CZ-0.5 and CZ-0.25, which is consistent with the trends in the I100/I002 ratio and the photocatalytic activity. The growth mechanism of the samples and the key factors determining the photocatalytic activity are discussed. Finally, the detailed surface characterization of the samples is described.
 Please wait while we load your content...
Please wait while we load your content...

