
Preparation of size controllable porous polymethylmethacrylate template and Cu micro/nanowire arrays†
Abstract
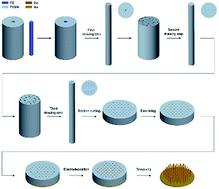
Polymethylmethacrylate (PMMA) composite fibers with micro/nanowire arrays of polystyrene (PS) fibers embedded were prepared by an iterative melt co-drawing and bundling technique. With section-cutting of the PMMA/PS fiber and the dissolution of inner PS, porous PMMA templates were obtained. The pore diameters and spacings of the template can be controlled by this method. The optical microscopy and scanning electron microscopy (SEM) pictures showed that the diameters of the pores were several microns and the spacings of pores were several tens of microns after the second drawing step. The diameters of the pores after the third drawing step were about 310 nm, and the spacings of pores were 2.30 μm. Then, size control of the Cu micro/nanowire arrays could be achieved by electrochemical deposition technology using this template. The SEM results indicated that the obtained diameters and spacings of the Cu micro/nanowires were in accordance with the size of the template. The obtained lengths of the Cu microwires could reach about 30 μm, which depended on the thickness of the PMMA template and parameters of the electrochemical deposition. Significantly, owing to the simple process and good controllability, various micro/nano array materials can be designed and constructed according to the application requirements by using this method.
 Please wait while we load your content...
Please wait while we load your content...

