
La–Mg mixed oxide as a highly basic water resistant catalyst for utilization of CO2 in the synthesis of quinazoline-2,4(1H,3H)-dione†

Abstract
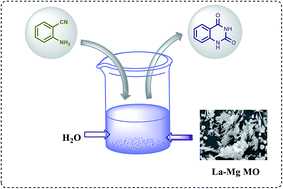
The synthesis of quinazoline-2,4(1H,3H)-dione was done by direct utilization of CO2 in the cyclization of 2-aminobenzonitrile (2-ABN) using lanthanum magnesia mixed oxide (La–Mg MO) as a strong basic catalyst under mild reaction conditions in water. It gave a conversion of ∼92% with 100% selectivity at 140 °C in 14 h. La–Mg MO was prepared by hydrothermal method using urea as homogeneous precipitating agent. The catalyst was characterized by different analytical techniques like BET, XRD, FT-IR, SEM, and TGA, and the basicity by CO2-TPD and acidity by NH3 TPD. Various reaction parameters were studied to predict the reaction mechanism and kinetics. The reaction follows the Langmuir–Hinshelwood–Hougen–Watson (LHHW) type kinetics model with an apparent activation energy of 23.3 kcal mol−1. The catalyst was recycled three times with an insignificant change in activity. The overall process is clean and green.
 Please wait while we load your content...
Please wait while we load your content...

