
Graphene enhanced low-density polyethylene by pretreatment and melt compounding†
Abstract
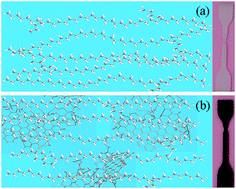
The rational design and fabrication of structural-functional materials are development trends in materials science. Herein, graphene enhanced low-density polyethylene (LDPE) is prepared by pretreatment and melt compounding, which is a simple and eco-friendly method. The amount of graphene added is controlled from 0 to 0.8 wt% to explore the enhancement effects. The graphene/LDPE nanocomposites (LPGNs) are characterized via SEM, TEM, Raman spectra, XRD, TG-DTA and DSC to research their dispersion morphology, crystal structure and thermal stability. A DMA, Servo Universal Strength Tester and Izod Impact Test Machine were used to study their mechanical properties. The results show that the added graphene is dispersed uniformly in the LDPE matrix, and the crystallinity of the LPGNs increases. The high specific surface area and outstanding properties of graphene improve the thermal stability, storage modulus, and mechanical properties of the LPGNs. Compared with neat LDPE, the Te of the LPGN with 0.8 wt% graphene increased by 58 °C and its tensile strength increased to 138%. A low content of graphene was effective in optimizing the Tm, Te and flame retardant properties of the LPGNs without compromising their mechanical properties.
 Please wait while we load your content...
Please wait while we load your content...

