
Structural and optical properties of sol–gel derived Cr-doped ZnO diluted magnetic semiconductor nanocrystals: an EXAFS study to relate the local structure†‡
Abstract
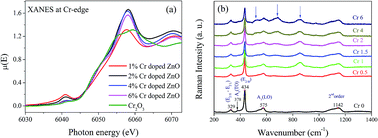
Structural, local structural and optical properties of sol–gel derived Zn1−xCrxO (0 ≤ x ≤ 0.06) nanoparticles have been thoroughly studied by several complementary techniques. The crystallite structure, size, and lattice strain have been estimated by X-ray diffraction (XRD) with Rietveld refinement and high-resolution transmission electron microscopy (HRTEM). No significant change in lattice parameters a and c has been observed upon Cr doping, though crystallite size and tensile strain in the crystals change. Extended X-ray absorption fine structure (EXAFS) measurement shows that Cr doping creates oxygen vacancies without causing any significant change in the host lattice structure. X-ray absorption near edge structure (XANES) measurements rule out the presence of metallic Cr clusters in the samples. Raman spectroscopy has been employed to study the crystalline quality, structural disorder, and defects in the host lattice. XANES and FTIR results show that the local structure around Cr becomes increasingly octahedral with an increase in Cr doping concentration. UV-vis measurements have been used to study the effect of Cr-doping on absorption spectra and hence on the band gaps of the samples. The band gap initially increases for low Cr-concentration and then decreases with higher Cr-concentration. The PL spectra show many emissions including green emission, which is attributed to singly ionized oxygen vacancies, increases with an increase in Cr doping concentration in the samples.
 Please wait while we load your content...
Please wait while we load your content...

