
Preparation of SDC–NC nanocomposite electrolytes with elevated densities: influence of prefiring and sintering treatments on their microstructures and electrical conductivities
Chuanming Li,
Yanwei Zeng*,
Zhentao Wang,
Zhupeng Ye,
Yuan Zhang and
Rui Shi
State Key Laboratory of Materials-oriented Chemical Engineering, School of Materials Science and Engineering, Nanjing Tech University, Nanjing 210009, PR China. E-mail: stephen_zeng@njtech.edu.cn; stephen_zeng@163.com; Fax: +86 25 83587254; Tel: +86 25 83587254
First published on 11th October 2016
Abstract
Sm0.2Ce0.8O1.9–Na2CO3 (SDC–NC) nanocomposite powders and electrolytes were prepared through the precipitation of Sm-doped cerium/sodium complex carbonate and its prefiring and sintering operations. Their phase components and microstructures were characterized by XRD, FT-IR, TG-DSC, SEM and TEM, respectively, and, in particular, the sintering performance and oxide ionic and protonic conductivities of SDC–NC nanocomposite electrolytes prepared by prefiring and sintering at different temperatures were studied. It has been found that the SDC–NC nanocomposite powders derived from pre-firing treatments of non-crystalline carbonate precipitates are composed of SDC/NC nanocomposite core–shell structured particles. Moreover, the as-sintered SDC–NC nanocomposite electrolytes are generally made up of densely compacted SDC particles bound by NC phase, while their sintering performances and microstructures are significantly affected by the prefiring and sintering temperatures due to the differences in structural homogeneity and continuity of the NC phase. In addition, the oxide ionic and protonic conductivities of SDC–NC nanocomposite electrolytes can be strongly dependent upon the prefiring and sintering treatments, with the sample S-500-800 (prefired at 500 °C and sintered at 800 °C) showing the highest conductivities, 9.11 and 3.27 mS cm−1 at 600 °C in H2 and air, respectively. The single cell based on the electrolyte of S-500-800 showed an OCV of 0.99 V and a peak power density of 342 mW cm−2 at 550 °C. More interestingly, the dependence of electrical performance on the prefiring and sintering temperatures is discussed from the perspective of the significant effects of the prefiring and sintering treatments on the microstructures and interfacial interactions between the phases of disperse SDC nanoparticles and NC, which is homogeneously and continuously filled in between them.
Introduction
Solid oxide fuel cells (SOFCs) have attracted growing attention due to their high energy conversion efficiency, environmentally friendliness and fuel flexibility.1–4 Traditional SOFCs based on yttrium-stabilized zirconia (YSZ) as the electrolyte have shown excellent performance, but often suffer the thermal degradation of materials for electrodes and electrolytes because of the high operation temperatures (800–1000 °C), which deteriorates the long-term stability and reliability of SOFC systems.5–10 Therefore, to reduce the cost of preparation and operation, a great deal of effort has been devoted to developing novel materials for electrodes and electrolytes which possess enough electrochemical activity in the intermediate temperature (IT) range (500–800 °C).In recent years, two-phase nanocomposite electrolytes composed of ceria-based oxides and alkali carbonate have been widely investigated as prospective electrolytes potentially applied to the intermediate temperature SOFCs.11–14 In these materials, the ceria-based oxides are usually rare-earth or alkali metal doped ceria such as Ce0.8Sm0.2O1.9 (SDC), Ce0.8Gd0.2O1.9 (GDC), Ce0.8Gd0.05Y0.15O1.9 (GYDC), Ce0.8Sm0.1Sr0.1O1.9 (SSDC) and Ce0.8Sm0.2−xCaxO2−d (CSDC),15–19 while the second phase is generally one of the Na, Li/Na or Li/K carbonates. Compared to the single phase SDC electrolytes, these doped ceria–carbonate nanocomposites have great advantages, such as concurrent conduction of protons and oxide ions as well as suppressed electronic conductivity in reducing atmosphere.20–24 As a result, the single cells with such nanocomposite electrolyte have been found to show excellent performance in the IT temperature range. B. Zhu et al. prepared a single cell with SDC–Li2CO3/Na2CO3 electrolytes and achieved a powder density of 200–800 mW cm−2 in the temperature range 400–600 °C.25 Y. Ma et al. realized a stable power density of 620 mW cm−2 over 12 hours on a single cell with SDC–NC nanocomposites.26 A single cell with SDC–NC nanocomposite electrolytes fabricated by X. D. Wang was reported to reach an output power density of 800 mW cm−2 at 600 °C and an OCV of 1.0 V.27 R. Raza et al. used CSDC–NC as electrolyte to work up single cells and obtained a power density of 980 mW cm−2 at 600 °C and 200 mW cm−2 at 350 °C with the OCV of 1.09 V and 0.99 V, respectively.28
However, in contrast to the good electrochemical performance of the single cells based on the SDC–carbonate composite electrolytes, their fairly low relative densities achieved by using light-sintering technologies, as reported in the literature,29–31 have become great concerns since such a low relative density is expected to cause the degradation of electrical performance and even the change in phase composition and microstructures during the long term operation of fuel cells. To improve the relative density, Y. F. Jing et al. applied the spark plasma sintering (SPS) method to the preparation of SDC–Li/Na carbonate nanocomposite electrolyte and found that the density may reach higher than 95% of the theoretical value. More importantly, the ionic conductivity of the SDC–Li/Na carbonate nanocomposite electrolyte treated with SPS was remarkably enhanced due to the improvement of microstructures.32 In addition, S. A. Muhammed et al. fabricated the SDC–Li/Na carbonate nanocomposite electrolyte with a relative density of 97% through optimizing the synthesis of SDC–carbonate nanocomposite powder and prolonging sintering time. Compared to the sample with a relative density of ∼85%, the single cell based on this highly densified SDC–Li/Na carbonate composite electrolyte demonstrated a 279% enhanced power density and 4% gained open circuit voltage, ∼63.3 mW cm−2 and 1.14 V at 650 °C, respectively.33 Evidently, an increase in relative density may remarkably improve the cell's performance in spite of the hitherto limited researches and, thus, more efforts should be committed to the densification study of SDC–NC composite electrolytes.
In this paper, we report our latest work on the SDC–NC nanocomposite electrolytes with elevated densities. Through the phase identification, microstructural characterization and the electrical conductivity measurements, the effects of the prefiring and sintering treatments on the microstructures and electrical performance of SDC–NC nanocomposite electrolytes have been systematically investigated. It is believed to be helpful for the deeper exploration and future development of high performance SDC–NC nanocomposite electrolytes.
Experimental procedure
Samples preparation
First of all, stoichiometric amounts of Ce(NO3)3·6H2O and Sm(NO3)3·6H2O were dissolved in deionized water to form a 0.5 M metal ions aqueous solution and then 0.5 M Na2CO3 aqueous solution as precipitant was added at a rate of 10 ml min−1 into the metal ions solution under vigorous stirring. After the precipitation was completed, the white precipitate of complex carbonate Na[Ce,Sm](CO3)2 (ref. 34) was separated through a vacuum filtration and washed three times with deionized water. It was then dried at 80 °C for 10 h before arranged to undergo prefiring at 400 °C, 500 °C and 600 °C for 2 h, respectively, to obtain the SDC–NC nanocomposite powders with the content of 46.8 vol% Na2CO3.To prepare the SDC–NC nanocomposite electrolytes, the as-prefired fluffy SDC–NC nanocomposite powders were ground in an agate mortar for 2 h with alcohol as a medium. Afterwards, the powders were pressed at 600 MPa into discs with dimensions Φ12 × 1 mm and then sintered at 700 °C, 750 °C and 800 °C for 5 h. According to the prefiring and sintering temperatures, the samples of SDC–NC nanocomposite electrolytes were designated by S-XX-YY, with XX and YY representing the prefiring and sintering temperature.
To fabricate the single cells for performance measurements, the as-sintered SDC–NC nanocomposite electrolyte pellets were abraded and polished carefully to 0.20 mm with 1200 mesh sandpaper. Then, the anode and cathode slurries were painted on the two side surfaces of each of the pellets to from the anode and cathode before they were subjected to a sintering at 700 °C for 1 h. Afterwards, the current collectors were made by coating Ag-paste on the cathodes and anodes to form the single cells with an active area of 0.52 cm2. In the meanwhile, the anode and cathode slurries were prepared by dispersing the composite anode/cathode materials, 40 wt% NiO/lithiated NiO and 60% SDC–NC nanocomposite powders in the solution of terpineol and ethyl cellulose.35–37
Characterization and electrical measurements
An X-ray diffractometer (ARL X'TRA) was used with Cu Kα radiation (λ = 1.5406 Å) and a tube power of 40 kV/35 mA for the phase identification. The morphology and microstructural features of the SDC–NC nanocomposite powders and as-sintered electrolytes were identified by SEM (Hitachi S-4800, Hitachi, Japan) and TEM (JEM-2100/JEOL, Japan). The relative density of the as-sintered samples was detected by Archimedean method as described in ASTM: B962-13.38,39 To trace the possible phase change within the precipitates, thermal analyses were performed on a TG-DSC (STA 449C, Netzsch, Germany) in the temperature range of 40–900 °C at a heating rate of 10 °C min−1 in air flow. Also, the infrared spectra of the precipitates were registered on a FTIR spectrometer (Nexus 670, Nicolet Co., USA) from 400 to 2400 cm−1.To study the electrical conductivities, the as-sintered electrolyte pellet samples were coated with Ag-electrodes on their two side faces. Their oxide ionic and protonic conductivity were measured in air and H2 (0.002 vol% H2O) atmosphere at the temperatures ranging from 400 to 600 °C, respectively, by AC impedance spectroscopy on an electrochemical workstation (Solartron SI-1260) with an alternating signal of 5 mV in the frequency range from 0.1 Hz to 1 MHz. As to the electrochemical performance tests of single cells, the measurements were carried out at 550 °C in the atmospheres of air on the side of cathode and H2 (0.002 vol% H2O) with a flow rate of 40 ml min−1 on the side of anode. The current–voltage curves were recorded on the electrochemical station (Solartron SI-1260).
Results and discussion
XRD, IR and TG-DSC diagrams of the dried precipitates
As shown in Fig. 1(a), the XRD pattern of the dried precipitate is featured with two extremely diffuse peaks around 2θ = 30° and 45°, indicating the presence of non-crystalline precipitate. However, from its infrared spectrum given in Fig. 1(b), the typical absorption bands for carbonate ions may be clearly identified, which are around 1062 cm−1, 1352 cm−1, 1400 cm−1 and 1456 cm−1, corresponding to their stretching vibrations, while the weak bands from 600 to 900 cm−1 arise from their bending vibrations.40–42 These results strongly suggest that the precipitate is composed of non-crystalline metal carbonates. Fig. 1(c) shows the typical TG-DSC curves of the dried precipitate. It can be seen that an endothermic peak appears at 131 °C along with a weight loss of 4.7% in TG curve, which may be attributed to the release of absorbed water. As the temperature is increased, an extended exothermic process with a sharp endothermic peak at 360 °C, accompanied by a drastic weight loss of 20.3%, should be associated with the release of crystallization water and the thermal decomposition of rare earth complex carbonate.43 As to the ramping DSC segment over 400 °C, it may be attributed to the endothermic reaction due to the improvement in crystallinity of SDC phase. Moreover, the total weight loss for the decomposition of carbonate precipitate is about 21% which is very close to that for the theoretical decomposition of SmCe–carbonate, 21.8%, strongly suggesting that the Na2CO3 phase maintain a stable state in the heat treatment process. | ||
| Fig. 1 XRD pattern (a), infrared spectrum (b) and TG-DSC curves (c) of the dried precipitate. | ||
Phase components and microstructures of SDC–NC nanocomposite powders
Fig. 2 shows the XRD patterns of SDC–NC nanocomposite powders prefired at 400 °C, 500 °C and 600 °C for 2 h, respectively. It can be seen that the diffraction peaks in each XRD pattern can be well matched with the data in JCPDS 75-0158 for crystalline SDC and no peaks may be found from crystalline NC phase and other impurity, regardless of some differences in the peak profiles. To track the presence of NC phase, the infrared spectra of the prefired SDC–NC composite powders were recorded and the typical one is given in Fig. 2(b). It can be found three infrared active vibration modes for crystalline NC phase consisting of the bands around 879 cm−1 for the polar vibration perpendicular to the plane of the ion, 1440 cm−1 and 700 cm−1 for the doubly-degenerate modes of asymmetric vibrations in the plane of the ion,44,45 indicating the existence of NC phase in SDC–NC nanocomposite powders. These results evidently imply that the SDC crystallite can be well formed after the non-crystalline carbonate precipitate is prefired at 400 °C or high temperatures for 2 h. More importantly, it also indicates that the NC phase is strongly prohibited from crystallization because of the strong interfacial interactions between the phases of SDC and NC.46,47 | ||
| Fig. 2 XRD patterns of the SDC–NC nanocomposite powders prefired at 400 °C, 500 °C and 600 °C (a), respectively, and their typical infrared spectrum (b). | ||
To determine the average grain sizes of the SDC phases in the different prefired samples, their XRD data were quantitatively analyzed with the help of Jade 5.0 software, with the best-fit results presented in Table 1. It can be seen that the cell parameters of SDC phases are of very close values, varying from 5.4327 Å to 5.4331 Å, but slightly larger than that of pure CeO2 (5.411 Å) due to the partial Sm3+ substitution for Ce4+ in CeO2. More interestingly, the average size of the SDC crystallites for the pre-fired samples shows a prominent increase from 7.6 to 19.2 nm as the pre-firing temperature is increased from 400 to 600 °C.
| Prefiring temperature (°C) | Lattice parameters (Å) | Average size (nm) |
|---|---|---|
| 400 | 5.4327 | 7.6 |
| 500 | 5.4329 | 9.6 |
| 600 | 5.4331 | 19.2 |
To characterize the microstructures of SDC–NC nanocomposite powders, FESEM observation was performed on the samples. Fig. 3 shows the typical FESEM images for the SDC–NC nanocomposite powders prefired at 400 °C, 500 °C and 600 °C, respectively. It can be seen that the particles in powders appear to be agglomerated and their average size is remarkably enlarged as the prefiring temperature is increased from 400 to 600 °C. To obtain further insight into the microstructural details, observation by TEM was carried out on the sample prefired at 600 °C. As shown in Fig. 3(d), the typical TEM image clearly demonstrates that the NC phase is tightly coated on the surface of SDC grains to form core–shell structured SDC/NC nanocomposite particles, in which the SDC crystallites act as cores and the shells are constituted by amorphous NC. Such a microstructural feature was ever reported in the literature19,27,34,46,48,49 and can be regarded as an evidence of the strong interfacial interactions between the phases of SDC and NC. So far, it can be concluded that the SDC–NC nanocomposite powders obtained through the prefiring treatments of non-crystalline complex carbonate precipitates are made up of SDC/NC nanocomposite particles, in which the SDC grain is tightly coated by the amorphous NC phase.
 | ||
| Fig. 3 FESEM images of SDC–NC nanocomposite powders prefired at 400 °C (a), 500 °C (b) and 600 °C (c) as well as TEM image of SDC–NC nanocomposite powder prefired at 600 °C (d). | ||
Phases and microstructures of sintered SDC–NC nanocomposites
In Fig. 4 is shown the XRD patterns of SDC–NC nanocomposites obtained at different prefiring and sintering temperatures, respectively. It can be found that all the XRD patterns are apparently the same as those of their prefired powders, suggesting that the SDC–NC nanocomposites are all composed of the same phase compositions: crystalline SDC and amorphous NC phases. Moreover, in accordance with the quantitative analysis of the XRD data, the SDC crystallite in these SDC–NC nanocomposites exhibit quite different average size, about ∼29.6, 31.2, 34.4, 44.5, 48.9, 50.9, 57.8, 60.4 and 67.5 nm in diameter according to their prefiring and sintering temperatures, respectively. Comparing with the increase of prefiring temperatures, it can be seen that the average size of SDC crystal shows a more prominent increase as the sintering temperature is increased from 700 to 800 °C, indicating that the SDC crystallites in SDC–NC nanocomposites have experienced a more significant growth in the sintering process than the prefired ones. | ||
| Fig. 4 XRD patterns of the SDC–NC nanocomposites obtained at different prefiring and sintering temperatures. | ||
Fig. 5 shows the relative density of all sintered SDC–NC nanocomposites as a function of sintering temperature. It can be found that the relative density of the samples prefired at the same temperature shows a prominent increase as the sintering temperature is increased from 700 to 800 °C and that of the samples sintered at the same temperature significantly decreases with the increase of prefiring temperatures. This may be because of that, on the one hand, the increased prefiring temperature is favorable for the agglomeration of SDC/NC nanocomposite particles and the presence of secondary particles, resulting in the decrease in the relative density for the sintered SDC–NC nanocomposites. On the other hand, in accordance with a higher sintering temperature is beneficial to the sintering densification, it is reasonable to find an increased relative density for the sintered SDC–NC nanocomposites with the increased sintering temperature. Hereinto, the S-600-700 sample gives the lowest relative density, approximately 71.5%, while the S-400-800 sample shows the highest, 92.8%, which may satisfy the requirements of IT electrolytes.
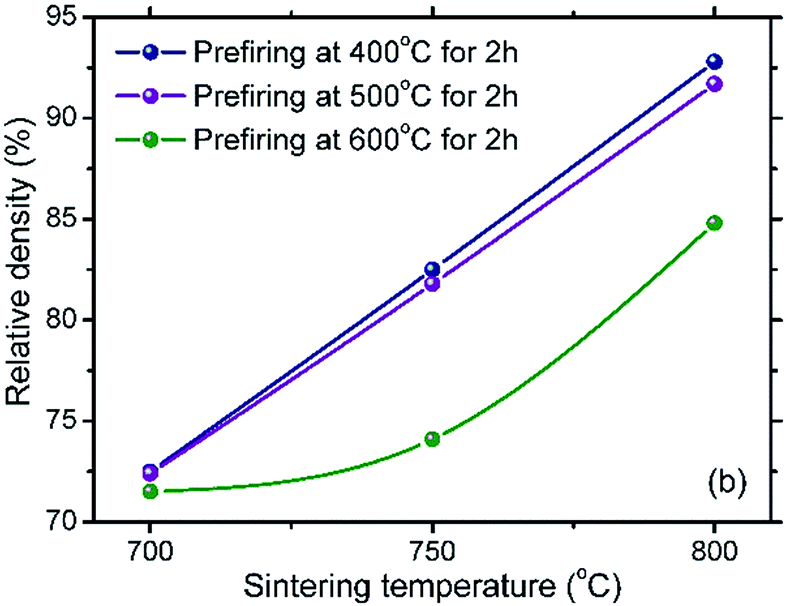 | ||
| Fig. 5 Relative density of SDC–NC nanocomposite samples obtained after prefiring and sintering at different temperatures. | ||
Fig. 6 illustrates the typical cross-sectional FESEM micrographs of the sample nanocomposites, and the packing voids are fully infiltrated with the NC phase in S-400-800 and S-500-800 while numerous of pores can be identified in S-600-800. Moreover, in contrast to the S-600-800, the NC phase in S-400-800 and S-500-800 shows a more homogenous and continuous distribution. In conclusion, a remarkably reduced sintering performance may be observed in the samples of S-400-800, S-500-800 and S-600-800 with the relative density decreased from 92.5% to 86.1%. To further explore the morphology and distribution of SDC particle, these three samples were soaked for 12 h in acetic acid (0.1 M) at room temperature so as to thoroughly remove the NC phase. As shown in Fig. 6(e)–(g), these three samples are all composed of loosely packed particles and a large number of pores in the samples can be well identified. To detect the phase components of these soaked samples, the EDS measurement was performed and the typical one was given in Fig. 6(g). It shows that only the elements belonging to SDC phase can be well identified in the as-soaked samples, indicating the NC phase has been removed completely. Therefore, it is reasonable to regard the pores in these as-soaked samples should be originally occupied by NC phase and correspondingly, it can be concluded that the relative density of sintered SDC–NC nanocomposites is apparently related to the distribution of NC phase and the higher the relative density of SDC–NC nanocomposite electrolyte is, the more homogeneity and continuity of the NC phase distributed of S-400-800, S-500-800 and S-600-800. First of all, it can be seen from Fig. 6(a)–(c) that a large number of particles are homogenously compacted inside the SDC–NC.
 | ||
| Fig. 6 Cross-sectional FESEM images of the samples of S-400-800 (a), S-500-800 (b) and S-600-800 (c) as well as the as-soaked samples S-400-800 (d), S-500-800 (e) and S-600-800 (f) and their typical EDS result (g). | ||
Electrical performance of sintered SDC–NC nanocomposites
Fig. 7 and 8 display the typical AC impedance spectra for the sample of S-500-800 measured in air and H2, respectively. It can be seen from Fig. 7(a) and 8(a) that the complex impedance spectra mainly consisting of several arcs express a significant alteration from the higher to lower resistance range as the measurement temperature is increased from 400 to 600 °C, suggesting a reduced electrolyte conductive resistance. In detail, as shown in Fig. 7(b) and 8(b), the AC impedance spectra obtained at 400 °C are composed of two parts: one is the arc in the high-frequency range, which is expected to be contributed by the grain/grain boundary effects and the other one is the part in the low frequency range may be determined by the electrode and/or mass diffusion process.50–52 As the measurement temperature increases from 400 to 600 °C, the arcs produced by the bulk resistance gradually degenerate and even disappear in some cases. Thus, in view of the above analysis, the equivalent circuit for the AC impedance spectra may be (Q1R1) (Q2R2) or R1 (Q2R2), in accordance with the different measuring temperatures, in which the overall resistance R1 represents the grain resistance and grain boundary resistance, and the pseudo-capacitance–resistance units Q2–R2 for the electrode processes. After the data-fitting analysis, the overall resistances R for each nanocomposite electrolyte at the different temperatures were converted into the oxide ionic and protonic conductivities in air and H2 in terms of the formula of σ = L/(RS), respectively, in which L is the thickness and S is the area of the as-measured sample. All of the ionic conductivities are represented in Fig. 9. | ||
| Fig. 7 Typical AC impedance spectra of S-500-800 measured in air at different temperatures from 400 to 600 °C (a) and the single ones at 400 °C (b) and 600 °C (c). | ||
 | ||
| Fig. 8 Typical AC impedance spectra of S-500-800 measured in H2 at different temperatures from 400 to 600 °C (a) and the single ones at 400 °C (b) and 600 °C (c). | ||
 | ||
| Fig. 9 Temperature dependence of oxide ionic conductivity (a) and protonic one (b) of SDC–NC nanocomposites obtained after prefiring and sintering at different temperatures. | ||
As can be seen from Fig. 9(a) and (b), the oxide ionic and protonic conductivities for the SDC–NC nanocomposites after prefiring and sintering at different temperatures are all exponentially increased as the temperature is increased from 400 to 600 °C, suggesting a working mechanism on the thermal activations. Moreover, compared with the oxide ionic conductivity of SDC–NC nanocomposites, the protonic conductivity is significantly enhanced. It should be noted that the reduction of Ce4+ into Ce3+ in H2 may be suppressed by the carbonate coating layer53 and then the electrical conduction for SDC–NC nanocomposites detected in H2 should be mainly contributed by the protons. Therefore, it is reasonable to regard the SDC–NC nanocomposites may be of the electrical conduction behavior that dominated by protons. Additionally, the sequence of the oxide ionic and protonic conductivities for these SDC–NC nanocomposites can be described as S-500-800 > S-400-800 > S-600-800 > S-500-750 > S-600-700 > S-400-750 > S-600-750 > S-500-700 > S-400-700. Hereinto, the highest oxide ionic and protonic conductivity is realized by the sample S-500-800, approximately 3.27 and 9.11 mS cm−1 at 600 °C. Indeed, as shown in Table 2, the previously reported oxide ionic conductivity is obviously smaller than that of the sample S-500-800, confirming an enhanced electrical conductivity.
As regards the fact that all SDC–NC nanocomposites are of the same phase compositions, but subjected to different prefiring and sintering temperatures, it is believed that the prefiring and sintering temperature should significantly affects the ionic conductivities of SDC–NC nanocomposites through influencing their interfacial interaction intensity34 and microstructures consisting of the average grain size of SDC crystals and the distribution of NC phase which is closely related to the relative density as we mentioned above. It can be understood that the higher intensity it is for the interfacial interactions, the larger conductivity the SDC–NC nanocomposites will be of. Moreover, as a definite consequence, one may expect that the SDC–NC nanocomposites which is with a smaller SDC nanocrystals and more continuous and homogenous distributed NC phase will prominently provoke a higher electrical conduction. Thus, in consideration of the sequence of oxide ionic and protonic conductivity is almost identical, the below section would be unfolded with the protonic conductivity as an example to analyze the relationships between these as-mentioned three factors and the electrical performance of SDC–NC nanocomposites obtained after prefiring and sintering at different temperatures.
Relationships between the protonic conductivity and the prefiring and sintering temperatures
In Fig. 10 shows the protonic conductivities of SDC–NC nanocomposites obtained after sintering at the same temperature. It can be seen from Fig. 10(a) that the protonic conductivity sequences for the SDC–NC nanocomposites sintered at 700 °C may be outlined as S-600-700 > S-500-700 > S-400-700. According to the similar average size of SDC nanocrystals, ∼29.6, 31.2 and 34.4 nm in diameter and relative density, approximately 72.5, 72.1 and 71.5%, the differences for these three samples in the protonic conductivity are supposed to be attributed to the interfacial interaction intensity between the phases of SDC and NC which may be summarized as S-600-700 > S-500-700 > S-400-700. This result is very comparable to the report of S. L. Yin34 and may further suggest an intensity sequences for the SDC–NC nanocomposite powder as S-600 > S-500 > S-400 due to the same sintering temperature. In addition, this result may determine that the intensity of the interfacial interactions play a key role in the electrical performance of the SDC–NC nanocomposite electrolytes when the samples possess a similar average size of SDC crystals and relative density. | ||
| Fig. 10 Temperature dependence of protonic conductivity of SDC–NC nanocomposites obtained after prefiring at different temperatures and sintering at 700 °C (a), 750 °C (b) and 800 °C (c). | ||
Moreover, it can be found in Fig. 10(b) and (c) that, in comparison with the SDC–NC nanocomposites sintered at 700 °C, the protonic conductivity order of that sintered at 750 °C and 800 °C may be shifted as S-500-750 > S-400-750 > S-600-750 and S-500-800 > S-400-800 > S-600-800, respectively. Compared with sample S-400-800, the sample S-500-800 has a similar mean grain size of the crystalline SDC and relative density (∼57.8/60.4 nm in diameter and 92.8/91.7%) but a higher protonic conductivity. This fact may further stress the importance of the interfacial interactions intensity for the electrical conductivity of the SDC–NC nanocomposites which possess a similar average size of SDC crystals and relative density. While, for sample S-600-800, in contrary to the highest interfacial interaction intensity, its largest average grain size of SDC crystals and lowest relative density are expected to tremendously decrease the contributions from the interfacial interactions across the phases of SDC and NC on the electrical conductivity of the SDC–NC nanocomposite electrolyte, resulting in the lowest protonic conductivity.
Fig. 11 gives the protonic conductivities of the SDC–NC nanocomposites prefired at the same temperature. As shown in Fig. 11(c), the protonic conductivity order for the SDC–NC nanocomposites prefired at 600 °C may be described as S-600-800 > S-600-700 > S-600-750. In accordance with the similar relative density for the sample S-600-700 and S-600-750, approximately 71.5/74.1%, their differences should be attributed to average grain size of SDC nanocrystals (34.4/50.9 nm in diameter) and the interfacial interactions intensity. Actually, according to the literature report,34 the interfacial interaction intensity may be prominently reduced as the heat treatment temperature is increased from 600 to 800 °C. Moreover, in comparison with the lowest electrical conductivity contribution from the average grain size of SDC nanocrystals and interfacial interactions intensity, the remarkably improved relative density, ∼84.8% for the sample S-600-800, may greatly modify the distribution of NC phase and lead to an enhanced protonic conductivity. More importantly, this result suggest that, compared with the mean grain size of SDC crystals and interfacial interaction intensity, the relative density may show the most pronounced influence on the electrical performance of SDC–NC nanocomposites through affecting the distribution of NC phase. Now let's take a look at Fig. 11(a) and (b). It gives the protonic conductivity order of the SDC–NC nanocomposites prefired at 400 and 500 °C which is followed by S-XX-800 > S-XX-750 > S-XX-700 and this result further demonstrate the importance of relative density for the electrical conductivity of the SDC–NC nanocomposites.
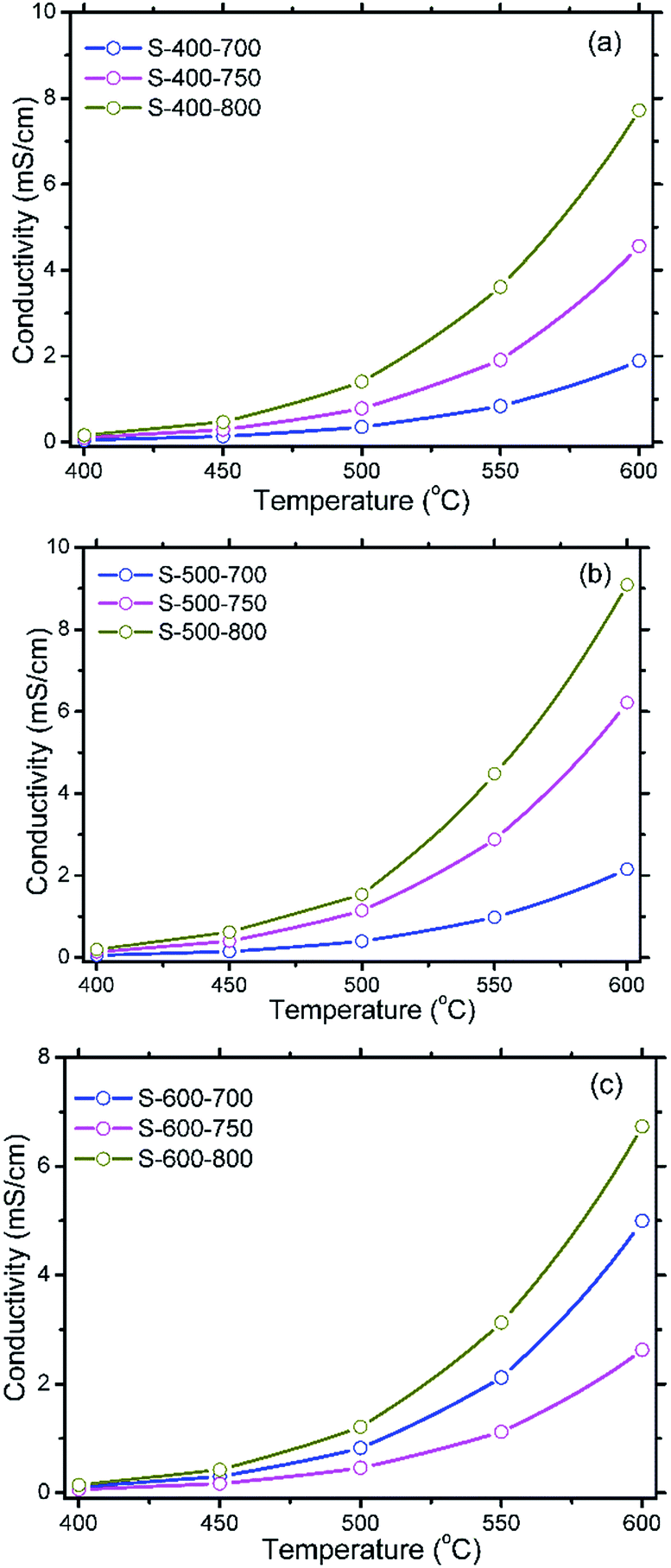 | ||
| Fig. 11 Temperature dependence of protonic conductivity of SDC–NC nanocomposites obtained after prefiring at 400 °C (a), 500 °C (b) and 600 °C (c) and sintering at different temperatures. | ||
Output performance of single cells
To further evaluate the influences from the relative density and oxide ionic and protonic conductivity on the electrical property of SDC–NC nanocomposites, performances of the single cell based on the elevated dense sample of S-400-800, S-500-800 and S-600-800 were given in Fig. 12. The open circuit voltage (OCV) of the three samples is about 1.002 V, 0.996 V and 0.961 V and the maximum power density reaches about 322, 341 and 268 mW cm−2 at 550 °C, respectively. These results show a good consistency with their relative densities and ionic conductivities. | ||
| Fig. 12 Output current/voltage/power density curves at 550 °C for the single cell based on the as-prepared SDC–NC nanocomposite electrolytes S-400-800, S-500-800 and S-600-800. | ||
Conclusions
In summary, according to the experimental results and related discussions on the relationships between the electrical performance and the prefiring and sintering temperatures, the following main conclusions can be drawn:(1) The SDC–NC nanocomposite powders derived from pre-firing treatments of non-crystalline carbonate precipitates are composed of SDC/NC nanocomposite core–shell structured particles. Moreover, the sintering performances and microstructures of the as-sintered SDC–NC nanocomposites are significantly affected by the prefiring and sintering temperatures due to the differences in structural homogeneity and continuity of NC phase.
(2) The oxide ionic and protonic conductivities of SDC–NC nanocomposites can be strongly dependent upon the prefiring and sintering treatments, with the sample S-500-800 showing the highest conductivities, 9.11 and 3.27 mS cm−1 at 600 °C in H2 and air, respectively. The single cell based on the electrolyte of S-500-800 showed an OCV of 0.99 V and peak power density of 342 mW cm−2 at 550 °C.
(3) The electrical performance of SDC–NC nanocomposite electrolytes were controlled by the prefiring and sintering treatments through affecting the microstructures consisting of the average size of SDC crystals and relative density and interfacial interactions between the phases of SDC and NC. In the meanwhile, the relative density may show the most pronounced influence by affecting the homogeneity and continuity of the NC phase.
Acknowledgements
The authors would like to gratefully acknowledge the funds by the Priority Academic Development Program of Jiangsu Higher Education Institutions, P. R. China, and the Program for Changjiang Scholars and Innovative Research Teams in Universities (PCSIRT) of China, IRT1146, and the Program of Research Innovation for University Graduate Students of Jiangsu Province (No. CXLX12_0427).References
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95 CrossRef CAS.
- V. Dusastre and J. A. Kilner, Solid State Ionics, 1999, 126, 163 CrossRef CAS.
- D. Stöver, H. P. Buchkremer and S. Uhlenbruc, Ceram. Int., 2000, 30, 1107 CrossRef.
- Y. Matsuzaki and I. Yasuda, Solid State Ionics, 2000, 132, 261 CrossRef CAS.
- M. F. Han, X. L. Tang, H. Y. Yin and S. P. Su, J. Power Sources, 2007, 165, 757 CrossRef CAS.
- Y. J. Leng, S. H. Chan, K. A. Khor and S. P. Jiang, Int. J. Hydrogen Energy, 2004, 29, 1025 CrossRef CAS.
- J. Liu and S. A. Barnett, Solid State Ionics, 2003, 158, 11 CrossRef CAS.
- S. Hui, J. Roller, S. Yick, X. Zhang, C. Deces-Petit, Y. Xie, R. Maric and D. Ghosh, J. Power Sources, 2007, 152, 493 CrossRef.
- S. C. Singhal and K. Kendall, High Temperature Solid Oxide Fuel Cells. Fundamentals, Design, and Applications, Elsevier Science Ltd, 2003 Search PubMed.
- B. Butza, P. Krusea, H. Störmera, D. Gerthsena, A. Müllerb, A. Weberb and E. Ivers-Tifféeb, Solid State Ionics, 2006, 177, 3275 CrossRef.
- B. Zhu and D. M. Mahmut, Int. J. Electrochem. Sci., 2006, 1, 383 CAS.
- T. X. Cai, Y. W. Zeng, S. L. Yin, L. Wang and C. M. Li, Mater. Lett., 2011, 65, 2751 CrossRef CAS.
- B. Zhu, R. Raza, Q. H. Liu, H. Q. Qin, Z. Q. Zhu, L. D. Fan, M. Singh and P. Lund, RSC Adv., 2012, 2, 5066 RSC.
- S. L. Yin, Z. P. Ye, C. M. Li, X. W. Chen and Y. W. Zeng, Mater. Lett., 2003, 92, 78 Search PubMed.
- L. D. Fan, G. Q. Zhang, M. M. Chen, C. Y. Wang, J. Di and B. Zhu, Int. J. Electrochem. Sci., 2012, 7, 8420 CAS.
- R. Chockalingam and S. Basu, Int. J. Hydrogen Energy, 2011, 36, 14977 CrossRef CAS.
- L. Zhang, R. Lan, X. Xu, S. W. Tao, Y. Z. Jiang and A. Kraft, J. Power Sources, 2009, 194, 967 CrossRef CAS.
- A. Rafique, R. Raza, N. Akram, M. K. Ullah, A. Ali, M. Irshad, K. Siraj, M. A. Khan, B. Zhu and R. Dawson, RSC Adv., 2015, 5, 86322 RSC.
- R. Raza, X. D. Wang, Y. Ma and B. Zhu, J. Power Sources, 2010, 195, 6491 CrossRef CAS.
- Y. Ma, X. D. Wang, H. A. Khalifa, B. Zhu and M. Muhammed, Int. J. Hydrogen Energy, 2012, 37, 19401 CrossRef CAS.
- J. B. Huang, F. C. Xie, C. Wang and Z. Q. Mao, Int. J. Hydrogen Energy, 2012, 37, 877 CrossRef CAS.
- J. B. Huang, Z. Q. Mao, L. Z. Yang and R. R. Peng, Electrochem. Solid-State Lett., 2005, 8, A437 CrossRef CAS.
- Q. H. Liu and B. Zhu, Appl. Phys. Lett., 2010, 97, 183115 CrossRef.
- Y. C. Zhao, C. Xia, Y. J. Wang, Z. R. Xu and Y. D. Li, Int. J. Hydrogen Energy, 2012, 37, 8556 CrossRef CAS.
- B. Zhu, J. Power Sources, 2003, 114, 1 CrossRef CAS.
- Y. Ma, X. D. Wang, R. Raza, M. Muhammed and B. Zhu, Int. J. Hydrogen Energy, 2010, 35, 2580 CrossRef CAS.
- X. D. Wang, Y. Ma, R. Raza, M. Muhammed and B. Zhu, Electrochem. Commun., 2008, 10, 1617 CrossRef CAS.
- R. Raza, H. Y. Qin, L. D. Fan, K. Takeda and M. Mizuhata, J. Power Sources, 2012, 201, 121 CrossRef CAS.
- Z. Gao, Z. Q. Mao, C. Wang, J. B. Huang and Z. X. Liu, Int. J. Energy Res., 2009, 33, 1138 CrossRef CAS.
- C. Xia, Y. Li, Q. H. Liu, Z. M. Wang, L. J. Jia, Y. C. Zhao and Y. D. Li, J. Power Sources, 2010, 195, 3149 CrossRef CAS.
- Y. C. Zhao, C. Xia, Z. R. Xu and Y. D. Li, Int. J. Hydrogen Energy, 2012, 37, 11378 CrossRef CAS.
- Y. F. Jing, Y. Ma, J. Patakangas, B. Zhu, M. Johnsson, M. E. Cura and P. Lund, Int. J. Hydrogen Energy, 2014, 39, 14391 CrossRef CAS.
- S. A. M. Ali, A. Muchtar, A. B. Sulong, N. Muhammed and E. H. Majlan, Ceram. Int., 2013, 39, 5813 CrossRef.
- S. L. Yin, Y. W. Zeng, C. M. Li, X. W. Chen and Z. P. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 12876 CAS.
- N. Ai, Z. Lü, K. F. Chen, X. Q. Huang, Y. W. Liu, R. F. Wang and W. H. Su, J. Membr. Sci., 2006, 286, 255 CrossRef CAS.
- C. J. Zhu, X. M. Liu, C. S. Yi, P. Li, D. J. Wang, D. T. Yan, K. G. Yao, T. Q. Lü and W. H. Su, J. Power Sources, 2010, 195, 3504 CrossRef CAS.
- X. D. Wang, Y. Ma, S. H. Li, B. Zhu and M. Muhammed, Int. J. Hydrogen Energy, 2012, 37, 19380 CrossRef CAS.
- K. S. Moon, H. Dong, R. Maric, S. Pothukuchi, A. Hunt, Y. Li and C. P. Wong, J. Electron. Mater., 2005, 34, 16 Search PubMed.
- T. Fruech, E. R. Kupp, C. Compson, J. Atria and G. L. Messing, J. Am. Ceram. Soc., 2016, 99, 2267 CrossRef.
- Y. Q. Zhai, S. Y. Zhang and H. Pang, Mater. Lett., 2007, 61, 1863 CrossRef CAS.
- H. S. Roh, H. Potdar, D. W. Jeong, K. S. Kim, J. O. Shim, W. J. Jang, K. Y. Koo and W. L. Yoon, Catal. Today, 2012, 185, 113 CrossRef CAS.
- E. J. Peterson and E. I. Onstott, J. Inorg. Nucl. Chem., 1979, 41, 517 CrossRef CAS.
- C. Padeste, N. W. Cant and D. L. Trimm, Catal. Lett., 1994, 24, 95 CrossRef CAS.
- M. J. Harris and E. K. H. Salje, J. Phys.: Condens. Matter, 1992, 4, 4399 CrossRef CAS.
- A. S. Al-Hawery, Jpn. J. Appl. Phys., 1998, 37, 858 CrossRef CAS.
- R. Raza, X. D. Wang, Y. Ma, X. R. Liu and B. Zhu, Int. J. Hydrogen Energy, 2010, 35, 2684 CrossRef CAS.
- C. M. Li, Y. W. Zeng, Z. T. Wang, F. Xu, Z. P. Ye and R. Shi, Electrochim. Acta, 2016, 212, 583 CrossRef CAS.
- X. D. Wang, Y. Ma and B. Zhu, Int. J. Hydrogen Energy, 2012, 37, 19417 CrossRef CAS.
- Y. Ma, X. D. Wang, H. A. Khalifa, B. Zhu and M. Muhammed, Int. J. Hydrogen Energy, 2012, 37, 19401 CrossRef CAS.
- A. S. V. Ferreira, C. M. C. Soares, F. M. H. L. R. Figueiredo and F. M. B. Marques, Int. J. Hydrogen Energy, 2011, 36, 3704 CrossRef CAS.
- B. Zhu, R. Raza, H. Y. Qin, Q. H. Liu and L. D. Fan, Energy Environ. Sci., 2011, 4, 2986 CAS.
- M. Benamira, A. Ringuedé, L. Hildebrandt, C. Lagergren, R. N. Vannier and M. Cassir, Int. J. Hydrogen Energy, 2012, 37, 19371 CrossRef CAS.
- J. T. Kim, T. H. Lee, K. Y. Park, Y. H. Seo, K. B. Kim, S. J. Song, B. Park and J. Y. Park, J. Power Sources, 2015, 275, 563 CrossRef CAS.
- C. M. Lapa, F. M. L. Figueiredo, D. P. F. de souza, L. Song, B. Zhu and F. M. B. Marques, Int. J. Hydrogen Energy, 2010, 35, 2953 CrossRef CAS.
- T. Ristoiu, T. Petrisor, M. Gabor, R. Simona, P. Florin, C. Traian and P. Traian, J. Alloys Compd., 2012, 532, 109 CrossRef CAS.
- A. Maheshwari and H. D. Wiemhöfer, Acta Mater., 2016, 103, 361 CrossRef CAS.
- F. Rahmawati, A. Nuryanto and K. D. Nugrahaningtyas, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 107, 012035 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |