
Coaxial ultrathin Co1−yFeyOx nanosheet coating on carbon nanotubes for water oxidation with excellent activity†
Yiyun Fang,
Xinzhe Li,
Shiling Zhao,
Juntian Wu,
Feng Li,
Min Tian,
Xuefeng Long,
Jun Jin* and
Jiantai Ma*
State Key Laboratory of Applied Organic Chemistry, The Key Laboratory of Catalytic Engineering of Gansu Province and Chemical Engineering, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: jinjun@lzu.edu.cn; majiantai@lzu.edu.cn; Fax: +86-931-8912582; Tel: +86-931-8912577
First published on 18th August 2016
Abstract
The rational design of high-performance and non-precious metal electrocatalysts is highly important for energy technologies. Herein, for the first time, a coaxial ultrathin Co1−yFeyOx nanosheet coating on carbon nanotubes (denoted as Co1−yFeyOx/CNTs) is prepared by a one-step pyrolysis method. The fabrication procedure is ultrafast and uncomplicated, furthermore, it does not need any reducing agent, alkali, or surfactant. In 1.0 M KOH, the obtained optimal hybrids Co0.8Fe0.2Ox/CNTs25 wt% catalyze oxygen evolution reactions (OER) with a very sharp onset potential (∼1.45 V) and an exceptional over-potential (0.28 V, at 10 mA cm−2) for more than 14 h, benefiting from the abundant active sites of ultrathin Co0.8Fe0.2Ox nanosheets, the hierarchical tubular architecture, and the strong cooperative effect among Co, Fe and CNTs. Remarkably, the excellent activity and durability of Co0.8Fe0.2Ox/CNTs25 wt% for the OER is superior to commercial RuO2 and many other highly active precious-metal/transition-metal catalysts reported to date. The design concept for tubular iron-group binary metal nanosheet hybrids creates new pathways for energy technologies.
Introduction
The increasingly serious energy crisis has caused extensive research into renewable energy, involving constructing innovative and high-performance energy storage equipment and exploiting earth-plentiful energy resources. Electrochemical water splitting is a feasible approach to acquire clean fuels from sustainable energy sources.1–3 Actually, the gross efficiency is extremely limited by the sluggish kinetics of the anodic OER process.4,5 Currently, the most highly efficient catalysts for the OER are RuO2 and IrO2, but even these operate with over-potentials exceeding 0.2 V at 10 mA cm−2.6 Moreover, the widespread application of Ru and Ir is constrained by their scarcity; thus the development of high-efficiency and inexpensive electrocatalysts based on earth-abundant and economic metals remains a crucial task.The iron-group composites have appeared as one of the most outstanding alternative electrocatalysts for OER owing to their considerable activity, earth-abundant, low cost, high durability as well as environmental friendliness.7–9 Plenty of previous researches demonstrate that a strong cooperative effect exists between iron-group and other metals, which can significantly increase the catalytic activity of the resulting binary metal catalysts.10–13 Most synthetic concepts for fabricating such catalysts for OER have been the co-precipitation of metal ions by an alkali, generally NH3·H2O or NaOH aqueous solution.14–18 Unfortunately, they do require to cautiously adjust the pH value of the solution for nanoparticles (NPs) formation and stabilization, and suffer from tremendously complicated and time-consuming preparation. Recently, an alternative approach to fabricate binary metal catalysts is organic phase decomposition of a precursor, for example, Yan et al. reported Au@Co3O4 core–shell nanocatalysts for OER through the reaction between Co(acac)2 (acac = acetylacetonate) and Au NPs in oleic acid and oleylamine.19 Although significant progress in fabricating binary metal catalysts has been made, it is required to use surfactant and reducing agent, which are difficult to separate from the obtained products. Besides, as far as we know, previous researches are mainly focused on preparing binary metal NPs as active sites in the past few years.17,19–22 Very recently, Xie and coworkers have found that the ultrathin nanosheets expose the largest number of electrochemical active sites, leading to a reduced diffusion path and an enhanced contact area with the solution.23–26 Thus, these kinds of materials are more appropriate alternatives for being high-performance catalysts. However, the use of iron-group binary metal nanosheets as electrocatalyst for OER is rarely reported up to now. For these reasons, it is desperately desirable yet a highly challenging to fabricate ultrathin iron-group binary metal nanosheets through a facile method with broad applicability as OER catalysts.
Iron-group metals usually have low intrinsical conductivity. To circumvent this issue, iron-group metals are generally loaded on a electroconductive surface to guarantee fast electron transfer. To our knowledge, CNTs are promising carriers for electrocatalysis owing to their superior properties.27–29 Unfortunately, most strategies to deposit active sites onto CNTs require chemical modification by introducing other functional groups, which could damage the sp2 carbon and reduce the electroconductivity of CNTs.30,31 Herein, for the first time, we fabricate a tubular iron-group binary metal nanosheet catalyst by a one-step procedure through the pyrolysis of Co(acac)2 and Fe(acac)3 in triethylene glycol (TREG) in the presence of CNTs. The obtained tubular hybrids Co1−yFeyOx/CNTs have the following advantages. Firstly, the fabrication procedure is ultrafast and uncomplicated, furthermore, it do not need any reducing agent, alkalis, and surfactant. Secondly, coaxial ultrathin Co1−yFeyOx exposes more electrochemical active sites than their bulk counterparts, resulting in a reduced diffusion path and an enhanced contact area with the solution. Thirdly, the obtained tubular hybrids have the hierarchical tubular architecture, where the most superficial layers supply abundant active sites for OER, while the internal walls maintain integrated and act as avenues for fast mass and electron transport. Most importantly, the Fe dopant in our catalysts can play a partial-charge-transfer-activation (PCTA) effect, resulting in enhanced the oxidation ability of CoIII/CoIV and increased the OER kinetics, which further leads to significant reduction of over-potentials and Tafel slope. Based on above reasons, the optimal Co0.8Fe0.2Ox/CNTs25 wt% hybrids demonstrate superior OER activity and durability compared with most of the high-performance electrocatalysts reported to date.
Experimental
Materials
High-purity (95%) multi-walled CNTs were purchased from Chengdu Organic Chemicals and used without further treatment. The outer diameters and lengths of the CNTs were 30–50 nm and 0.5–2 μm, respectively. Co(acac)2 and Fe(acac)3 was purchased from Energy Chemical Reagent Co., Ltd. RuO2 (99.95%, metal basis) were purchased from Alfa Aesar. All chemicals used in this work were of analytical grade and used as supplied.Synthesis of Co1−yFeyOx/CNTs composites
The tubular Co1−yFeyOx/CNT hybrids were synthesized through a pyrolysis process. Typically, Co(acac)2 (32.1 mg, 0.0899 mmol) and Fe(acac)3 (7.9 mg, 0.0225 mmol) were added to TREG (15 mL), next the obtained compound was ultrasonicated to form a well-mixed solution (ensure the total mass of Co(acac)2 and Fe(acac)3 always remains 40 mg). Then, different amounts of CNTs (15, 20, 25, 30 and 35 wt%) were added to the mixtures based on the starting total mass of Co(acac)2 and Fe(acac)3. Afterwards, the mixture was ultrasonicated for another 10 min, followed by heating to 278 °C at reflux for 0.5 h under Ar flow. After cooling to ambient temperature, the obtained materials were filtered, washed with distilled water and ethanol several times. Finally, the obtained materials were dried in a vacuum oven for 24 h. The weight percentages of Co and Fe in the obtained product are defined by inductive coupled plasma atomic emission spectrometer (ICP-AES) analysis, conducted with Perkin Elmer (Optima-4300DV), was 28.7% and 1.53%, respectively. The molar ratio (Co/Fe) in obtained materials was regulated by altering the primal mass of Co(acac)2 and Fe(acac)3.Synthesis of physically mixed Co0.8Fe02Ox with CNTs
The pure Co0.8Fe0.2Ox composites were prepared by the directly pyrolysis of Co(acac)2 (32.1 mg) and Fe(acac)3 (7.9 mg) in triethylene glycol (15 mL) without addition of CNTs. Then the obtained Co0.8Fe0.2Ox were physically mixed with CNTs to obtain physically mixed Co0.8Fe0.2Ox + CNTs. The content of Co and Fe in mixed Co0.8Fe0.2Ox + CNTs was adjusted to be equal to the percentages in Co0.8Fe0.2Ox/CNTs25 wt%.Electrochemical measurements
The electrochemical measurements were implemented in a three-electrode electrochemical cell, in which glassy carbon electrode (GCE, diameter: 3.0 mm) served as working electrode, 3.0 M KCl–Ag/AgCl served as reference electrode, and Pt wire served as counter electrode. The preparation of GCE was carried out as hereunder mentioned: the GCE was polished with Al2O3 to acquire a clean surface. Then, the fabricated catalysts (5 mg) and 0.5 wt% Nafion solution (100 μL) were added into ethanol/water solvent (1 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) with slightly ultrasonicated to obtain a homogenous catalyst ink. Next, the suspensions (8 μL) was dripped onto the GCE surface and fully dried in air (catalysts loading ∼0.52 mg cm−2). The OER performance of all materials were studied by the linear sweep voltammogram (LSV) in 1.0 M KOH at the scan rate of 2 mV s−1. O2 was purged into the electrolyte to attain an O2-saturated condition. Electrochemical impedance spectroscopy (EIS) were measured from 105 to 0.1 Hz with an AC voltage (5 mV) in O2-saturated KOH (1.0 M). Durability tests was performed by means of chronopotentiometry (CP) and chronoamperometry (CA) measurements. The potentials were converted to the reversible hydrogen electrode (RHE) scale using eqn: ERHE = EAg/AgCl + 0.059 × pH + EoAg/AgCl (EoAg/AgCl = 0.198).
:1 v/v) with slightly ultrasonicated to obtain a homogenous catalyst ink. Next, the suspensions (8 μL) was dripped onto the GCE surface and fully dried in air (catalysts loading ∼0.52 mg cm−2). The OER performance of all materials were studied by the linear sweep voltammogram (LSV) in 1.0 M KOH at the scan rate of 2 mV s−1. O2 was purged into the electrolyte to attain an O2-saturated condition. Electrochemical impedance spectroscopy (EIS) were measured from 105 to 0.1 Hz with an AC voltage (5 mV) in O2-saturated KOH (1.0 M). Durability tests was performed by means of chronopotentiometry (CP) and chronoamperometry (CA) measurements. The potentials were converted to the reversible hydrogen electrode (RHE) scale using eqn: ERHE = EAg/AgCl + 0.059 × pH + EoAg/AgCl (EoAg/AgCl = 0.198).
Characterizations
A transmission electron microscope (FEI TecnaiTM F30, USA, 300 kV) was used to make the transmission electron microscopy (TEM) images, high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM), high resolution TEM images (HRTEM). Linear distribution of composition elements and elemental mapping was obtained through the energy dispersive X-ray spectroscopy (EDS, AMETEK, USA) attached to the transmission electron microscope. The synthesized samples suspension for analysis was dropped onto an ultrathin carbon-coated holey carbon film holder with 300 mesh copper grid and fully dried at room condition. Scanning electron microscopy (SEM, Hitachi S-4800, Chiyoda-ku, Tokyo, Japan) analysis was measured by dropping the ethanol solution containing the catalyst sample onto the to the silicon substrate. X-ray diffraction (XRD) was measured on the Rigaku D/max-2400 diffractometer using a Cu Kα X-ray. X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALAB210 spectrometer (VG, UK). Raman spectra were recorded on a Jobin-Yvon LabRam HR80 spectrometer (Horiba Jobin Yvon, Inc.) with a laser source of 532 nm. The weight percentages of Co and Fe in catalysts are measured on by ICP-AES analysis, conducted with Perkin Elmer (Optima-4300DV).Results and discussion
The preparation concept for tubular Co1−yFeyOx/CNTs hybrid electrocatalysts through the pyrolysis of Co(acac)2 and Fe(acac)3 in TREG in the presence of CNTs was illustrated in Fig. 1. Briefly, Co(acac)2, Fe(acac)3 and CNTs were ultrasonicated in TREG to form a uniform solution, followed by heating to 278 °C at reflux for 0.5 h under Ar flow, yielding Co1−yFeyOx/CNTs catalysts.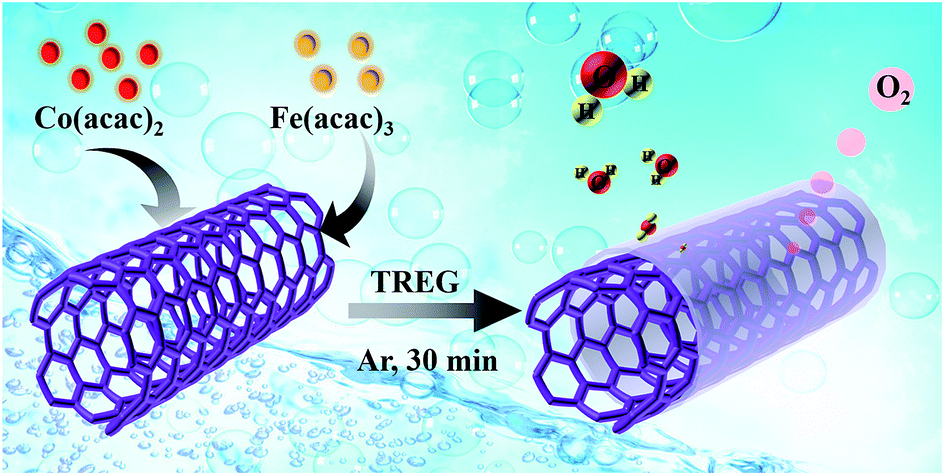 | ||
| Fig. 1 Schematic illustration of the synthesis of Co1−yFeyOx/CNTs hybrids. | ||
The morphology and microstructure of the synthesized Co0.8Fe0.2Ox/CNTs25 wt% hybrids could be seen from the TEM, HRTEM, HAADF-STEM, SEM, and elemental mapping images. As shown in Fig. 2a, the pure CNTs without introducing other groups for functionalization are intact, retaining their original smooth sidewall and tubular structure. After pyrolysis of Co(acac)2 and Fe(acac)3 in TREG in the presence of CNTs, coaxial ultrathin Co0.8Fe0.2Ox nanosheets coating on the surfaces of CNTs are obtained. TEM image of Co0.8Fe0.2Ox/CNTs25 wt% in Fig. 2b reveals that the tubular architecture of CNTs does not be destroyed during pyrolysis process, and the most superficial layers of CNTs become coarse after coating coaxial Co0.8Fe0.2Ox nanosheet. Image in Fig. 2c illustrates the same characteristics. In order to explore the thickness of Co0.8Fe0.2Ox nanosheets, HRTEM image is taken and showed in Fig. 2d. The Co0.8Fe0.2Ox nanosheets is so ultrathin and intact that the appearance of tubular structure nearly unaltered except getting coarse. The thickness of Co0.8Fe0.2Ox nanosheets is about 2 nm. From HRTEM image, we can also clearly see the multi-layered structure of the Co0.8Fe0.2Ox/CNTs25 wt% catalysts. Thanks to the multi-layered structures of CNTs, this novel architecture not only makes the outermost layers Co0.8Fe0.2Ox supply abundant active sites for OER, but also ensure the internal walls maintain integrated and act as avenues for fast mass and electron transport. Upon loading Co0.8Fe0.2Ox nanosheet, the whole resulting Co0.8Fe0.2Ox/CNTs25 wt% catalysts inherit the overall tubular morphology (Fig. 2e), which also can be observed from HAADF-STEM (Fig. 2f). By EDS analysis (inset Fig. 2e), it can be concluded that the Co0.8Fe0.2Ox/CNTs25 wt% hybrids contain C, O, Co, Fe and Cu. Cu is generally derived from the copper grid, C, O, Co and Fe signals result from the Co0.8Fe0.2Ox/CNTs25 wt%. The linear distribution of chemical elements composition of monoradicular Co0.8Fe0.2Ox/CNTs25 wt% catalyst (Fig. 2g, red line) is also studied by the EDS, and the results indicate that the Co and Fe are evenly distributed on the Co0.8Fe0.2Ox. The corresponding elemental mapping results also confirm the homogenous distribution of C, O, Co and Fe elemental species in the Co0.8Fe0.2Ox/CNTs25 wt% (Fig. 1g). SEM and the corresponding EDS mapping characterization illustrate the same conclusion (Fig. S1†).
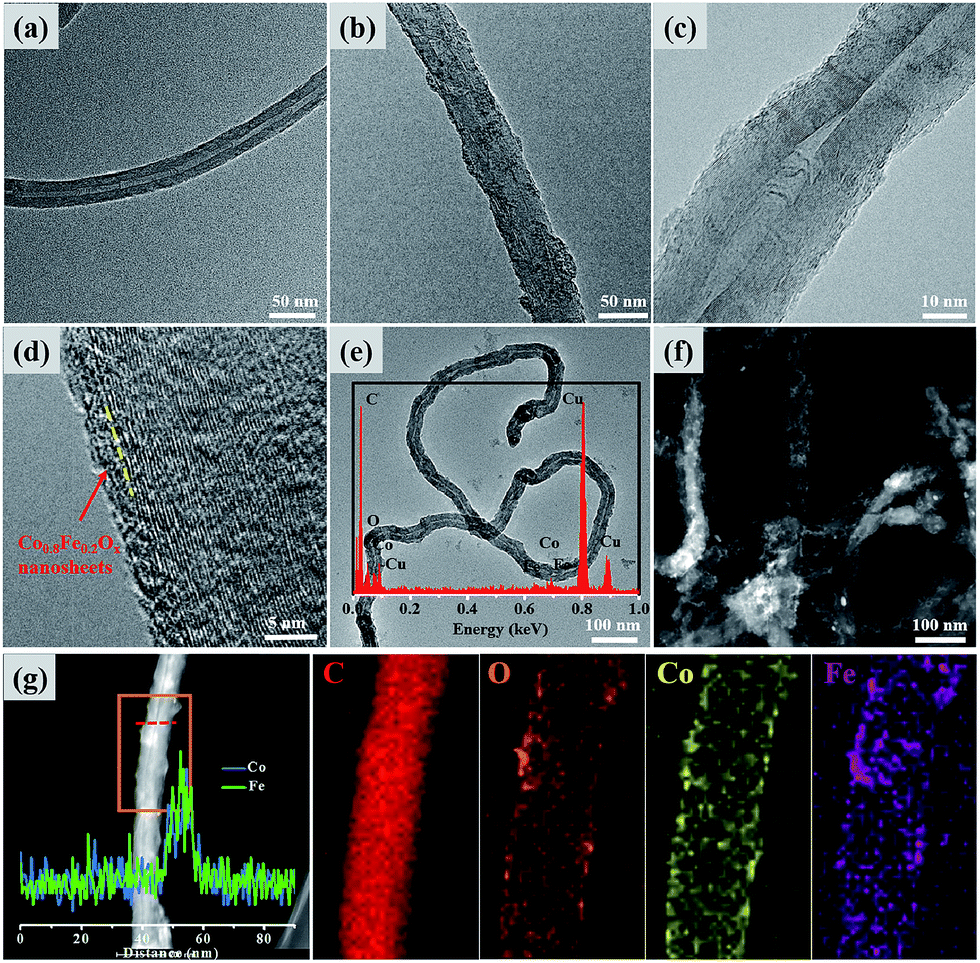 | ||
| Fig. 2 TEM images of (a) pure CNTs, (b, c and e) Co0.8Fe0.2Ox/CNTs25 wt%. HRTEM (d) of Co0.8Fe0.2Ox/CNTs25 wt% (inset (e): EDS spectrum of the catalysts). Typical HAADF-STEM image of (f) Co0.8Fe0.2Ox/CNTs25 wt%. (g) HAADF-STEM image of Co0.8Fe0.2Ox/CNTs25 wt% and the corresponding EDS mapping. | ||
XRD was used to identify the crystal phases and structure of the as-prepared products. As shown in Fig. 3a, the diffraction peaks at 26° can be attributed to the graphite structure (002) plane of the CNTs.32–34 For pure Co(acac)2 precursors, the obtained products Co/CNTs25 wt% have the hexagonal closed-packed (hcp) phase of metallic Co (JCPDS no. 01-1254). However, on addition of Fe(acac)3 (Co/Fe = 4:1), clear signals for spinel compounds appear, suggesting that two compositions exist, the spinel Fe–Co–O and metal. Interestingly, the crystal phase is changed to face-centered cubic (fcc) structure (JCPDS no. 01-1255) from the hcp structure, which may result from the formation of the spinel CoFe2O4.35 After the thermal decomposition process only with Fe(acac)3, a few new peaks are observed. The positions and relative intensities of these non-CNTs related new peaks match well with the planes of the standard XRD data for Fe3O4 (JCPDS no. 19-0629), which confirms the formation of products Fe3O4/CNTs25 wt%.28,36 Therefore, the resulting products can be greatly affected by the precursor composition. In addition, no shift in peak position was observed for the spinel, indicating that different iron doping levels do not affect the phase of spinel product.
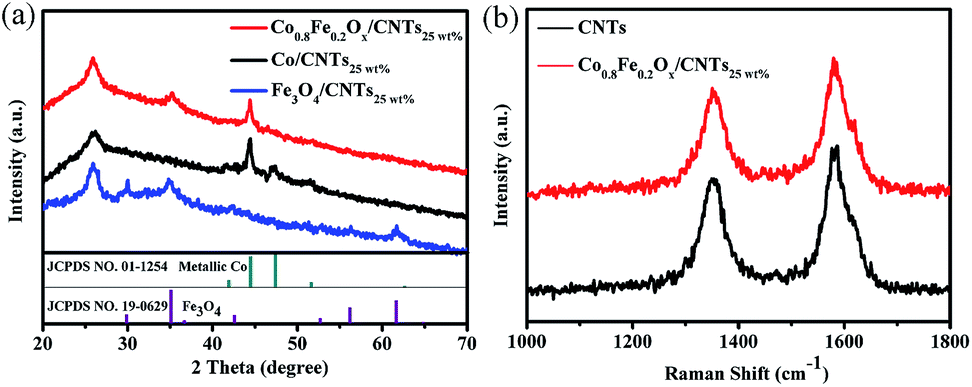 | ||
| Fig. 3 (a) Wide-angle XRD patterns of Fe3O4/CNTs25 wt%, Co/CNTs25 wt% and Co0.8Fe0.2Ox/CNTs25 wt% composites. The standard XRD pattern of Fe3O4 (JCPDS no. 19-0629) and Co (JCPDS no. 01-1254) are shown beneath the plots. (b) Raman spectra of CNTs and Co0.8Fe0.2Ox/CNTs25 wt%. | ||
Raman spectroscopy was employed to investigate the characteristic D (∼1320 cm−1) and G (∼1570 cm−1) bands of the CNTs and Co0.8Fe0.2Ox/CNTs25 wt%, and the result was shown in Fig. 3b. The D band is attributed to the hybridized vibrational mode related to the defects and edges of CNTs. The G band is due to the tangential oscillation and vibration from sp2 carbon in CNTs.34,37 Hence, the ID/IG value is usually applied to estimate the defect density in CNTs. In Co0.8Fe0.2Ox/CNTs25 wt% hybrids, ID/IG is 0.87, higher than that of CNTs (0.78), which means that the ultrathin Co1−yFeyOx nanosheets can bring more microstructural rearrangement or structural defects to the CNTs. Plausibly, such defects from Co0.8Fe0.2Ox can help to improve the catalytic performance owing to the improved π-bonding and electron donor–acceptor features.33,34,37,38
XPS characterization was then carried out to confirm the chemical constitution and the chemical bonding of constituent elements in Co0.8Fe0.2Ox/CNTs25 wt%. As shown in Fig. 4a, Co 2p, Fe 2p, O 1s and C 1s peaks are found in the Co0.8Fe0.2Ox/CNTs25 wt% sample. Moreover, two pairs of Co peaks are observed in Fig. 4c, where the peaks at 780.8 (Co 2p3/2) and 796.4 eV (Co 2p1/2) derive from CoFe2O4, while the peaks at 785.6 (Co 2p3/2 sat) and 803.0 eV (Co 2p1/2 sat) are due to the Co(OH)2, instead of metallic Co.35 The reason is that when these active Co are exposed to air (containing O2 and H2O vapor), they are oxidized to Co(OH)2.35 The Fe 2p spectra shows two peaks at about 710.8 eV and 724.3 eV (Fig. 4d), corresponding to Fe 2p3/2 and Fe 2p1/2, indicating the exist of Fe3+.15 In comparison to the XPS peak at 781.1 eV attributed to Co 2p3/2 of Co/CNTs25 wt%, and 710.4 eV attributed to Fe 2p3/2 of Fe3O4/CNTs25 wt% (Fig. S2†), the shift of the corresponding peaks imply the great coactions between Co and Fe. To accurately manifest the specific interaction between the CNTs and Co1−yFeyOx, XPS characterizations of Co0.8Fe0.2Ox and Co0.8Fe0.2Ox/CNTs25 wt% were carried out and compared. As shown in Fig. S3,† in comparison to the XPS peak centered at 780.2 eV assigned to Co 2p3/2 of Co0.8Fe0.2Ox, the shift of the corresponding peak of Co0.8Fe0.2Ox/CNTs25 wt% to 780.8 eV implies the close assembly and strong interaction between Co0.8Fe0.2Ox/CNTs25 wt% and CNTs, resulting in the impaired electron density of Co atoms in Co0.8Fe0.2Ox/CNTs25 wt%.
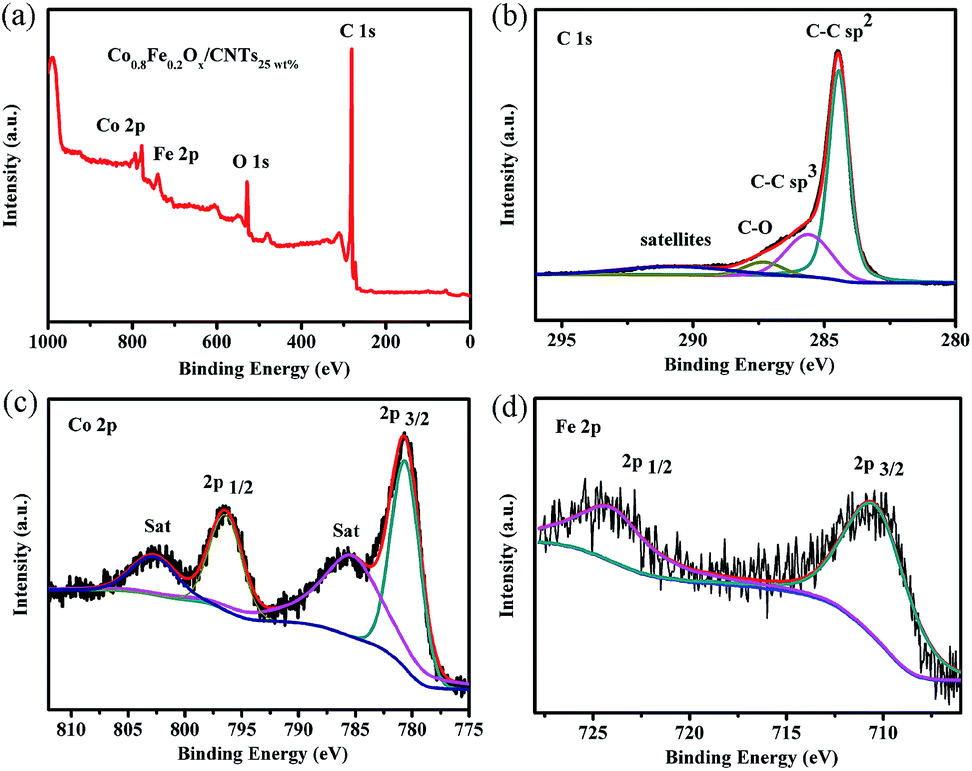 | ||
| Fig. 4 (a) XPS wide-scan spectrum, (b) deconvoluted C 1s spectrum, (c) deconvoluted Co 2p spectrum and (d) deconvoluted Fe 2p spectrum of Co0.8Fe0.2Ox/CNTs25 wt% composites. | ||
The OER activities of Co0.8Fe0.2Ox/CNTs25 wt% hybrids and other contrast samples were assessed in O2-saturated 1.0 M KOH by LSV at scan rate of 2 mV s−1 at room temperature. As expected (Fig. 5a), Co0.8Fe0.2Ox/CNTs25 wt% demonstrates a sharp onset potential (∼1.45 V). As contrasts, monocomponent Co/CNTs25 wt% and Fe3O4/CNTs25 wt% hybrids are also performed under the same condition. It is observed that Co/CNTs25 wt% and Fe3O4/CNTs25 wt% show obviously higher onset potential (∼1.53 and ∼1.60 V, respectively), manifesting the Fe impurity can obviously improve the OER performance. An another criterion to assess the OER performance is the Tafel slope (b), which is obtained by fitting linear portion of Tafel data to the Tafel equation (η = a + blog|J|, where η is the over-potential, b is the Tafel slope, and J is the current density). To achieve more insight to the OER process of those catalysts, the relevant Tafel slope is obtained. As revealed in Fig. 5b, the Tafel slope value of the Co0.8Fe0.2Ox/CNTs25 wt% catalyst is ∼49 mV dec−1, far less than CNTs (∼190 mV dec−1), Co/CNTs25 wt% (∼81 mV dec−1) and Fe3O4/CNTs25 wt% (∼98 mV dec−1), which further indicates that Co0.8Fe0.2Ox/CNTs25 wt% composites are more desirable for OER than the single metal materials. Besides the onset potential and Tafel slope, the over-potential required for the current density of 10 mA cm−2 (defined as η0) is commonly used as another important parameter to assess the OER performance,34,39 and analogous trend can be detected by employing this criterion. Particularly, the η0 of Co0.8Fe0.2Ox/CNTs25 wt% catalysts is only 0.28 V, which is the lowest among all materials (Fig. 5c), e.g., 0.38 V for Co/CNTs25 wt%, 0.43 V for Co0.8Fe0.2Ox and 0.47 V for Fe3O4/CNTs25 wt%. As a consequence, appropriate Fe doping leads to significant decreases of Tafel slope and over-potentials. As for pure CNTs, the OER current can not attain 10 mA cm−2 even at η0 = 0.47 V. In addition, the contribution of substrate CNTs is revealed. As exhibited in Fig. 5d, the onset potential is shifted by about 0.1 V for Co0.8Fe0.2Ox catalysts compared with Co0.8Fe0.2Ox/CNTs25 wt% catalysts, and the Tafel slope and η0 are as high as ∼85 mV dec−1 and 0.43 V, respectively. By adjusting the ratio of CNTs to the total mass of starting Co(acac)2 and Fe(acac)3, we find that Co0.8Fe0.2Ox/CNTs25 wt% exhibit the best OER activity among Co0.8Fe0.2Ox/CNTs15 wt%, Co0.8Fe0.2Ox/CNTs20 wt%, Co0.8Fe0.2Ox/CNTs25 wt%, Co0.8Fe0.2Ox/CNTs30 wt% and Co0.8Fe0.2Ox/CNTs35 wt% (Fig. 5d). The reason is redundant CNTs would decrease the proportion of active sites in catalysts and repress OER performance.40 A particular contrast of many high-performance materials for OER was presented in Table S1,† further proving the excellent OER activity is superior to many other highly active precious-metal/transition-metal catalysts reported to date.
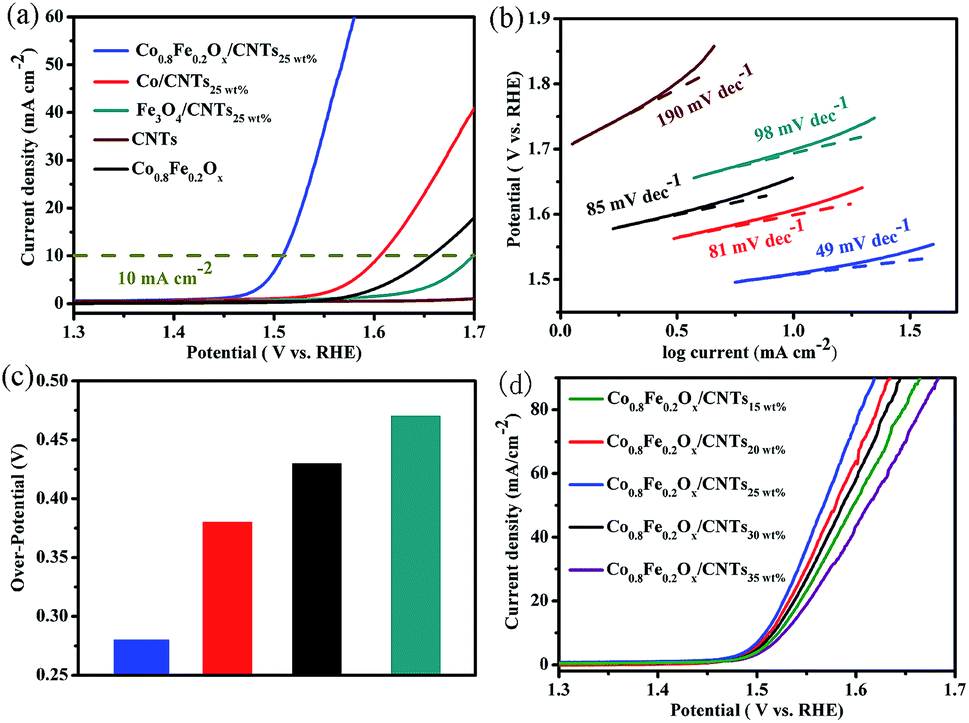 | ||
| Fig. 5 (a) LSV curves of CNTs, Fe3O4/CNTs25 wt%, Co/CNTs25 wt%, Co0.8Fe0.2Ox and Co0.8Fe0.2Ox/CNTs25 wt% in N2-saturated 1.0 M KOH solution (scan rate 2 mV s−1). (b) Tafel plots of catalysts above. (c) Comparison of the over-potential for different catalysts at the current density of 10 mA cm−2. (d) LSV curves of Co0.8Fe0.2Ox/CNTs15 wt%, Co0.8Fe0.2Ox/CNTs20 wt%, Co0.8Fe0.2Ox/CNTs25 wt%, Co0.8Fe0.2Ox/CNTs30 wt% and Co0.8Fe0.2Ox/CNTs35 wt% in 1.0 M KOH solution. | ||
The LSV curves of various proportions of Co and Fe in Co1−yFeyOx/CNTs25 wt% were measured in the same system. The weight percentage of CNTs to starting total mass of Co(acac)2 and Fe(acac)3 is kept constant (25 wt%) for this assessment. As displayed in Fig. 6a, the Co0.80Fe0.20Ox/CNTs25 wt% catalysts not only show a more sharp onset potential, but also improve kinetics for OER, which is demonstrated by its lower Tafel slope of ∼49 mV dec−1 contrasted with other catalysts, e.g., Co0.67Fe0.33Ox/CNTs25 wt% (∼58 mV dec−1), Co0.75Fe0.25Ox/CNTs25 wt% (∼51 mV dec−1), Co0.83Fe0.17Ox/CNTs25 wt% (∼56 mV dec−1) and Co0.86Fe0.14Ox/CNTs25 wt% (∼74 mV dec−1), as reveled in Fig. 6b. Similar to the previous discoveries that Fe doping can significantly improve the catalytic performance of iron-group composites, the Co1−yFeyOx/CNTs also reveal constituent-determined activity. It can be clearly observed that both Tafel slope and current density display a “volcano”-type relation versus Co/Fe ratio, and the best value is 4:1. Corrigan and Boettcher's research illustrated that Fe could play a PCTA effect and act as very effective activators.41,42 In this work, the PCTA effect also devotes to enhancing the OER performance. It should be noted that CoIV has been testified as the active centres for the OER,9,11,19,43,44 and the higher oxidation potential of CoIV/CoIII, the stronger oxidizing ability of catalysts. For Co1−yFeyOx/CNTs catalysts, FeIII is predominant. When the FeIII is doped into the materials, PCTA effect occurs, and then FeIII is oxidized to FeIV.41,42,45 The transferred electrons during this process could restrain the oxidation from CoII to CoIII and result in less amount of CoIII, which further lead to the higher oxidation potential of CoIV/CoIII and stronger oxidizing ability of Co1−yFeyOx/CNTs catalysts.35,46
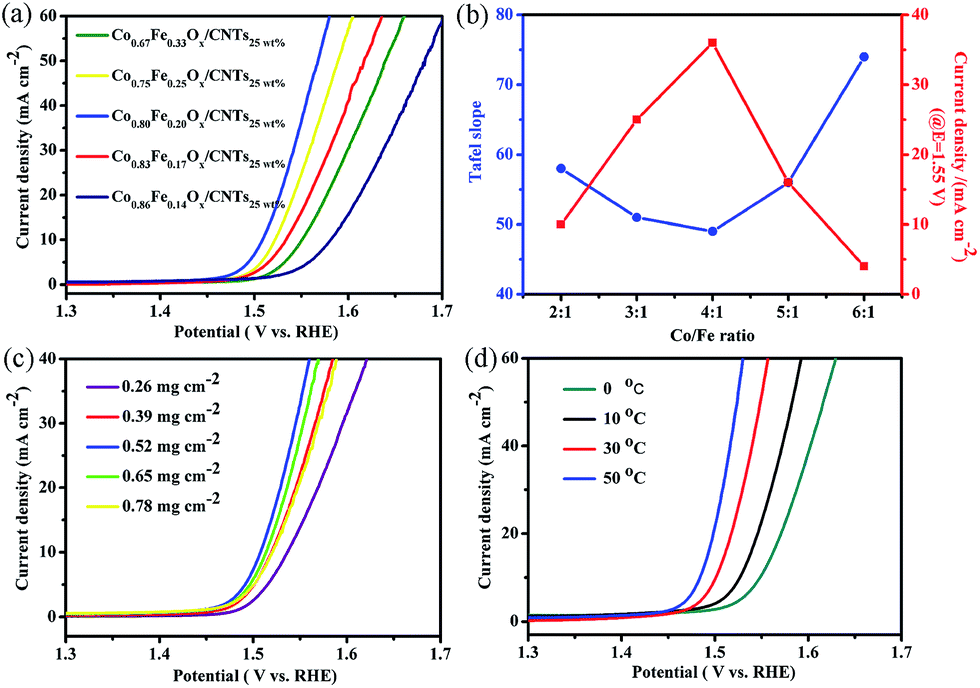 | ||
| Fig. 6 (a) LSV curves of Co0.67Fe0.33Ox/CNTs25 wt%, Co0.75Fe0.25Ox/CNTs25 wt%, Co0.80Fe0.20Ox/CNTs25 wt%, Co0.83Fe0.17Ox/CNTs25 wt% and Co0.86Fe0.14Ox/CNTs25 wt% in 1.0 M KOH solution. (b) Co/Fe ratio-dependent Tafel slope plots and over-potential (10 mA cm−2) of the products. (c) LSV curves of the Co0.80Fe0.20Ox/CNTs25 wt% on GCE at five different mass loadings. (d) LSV curves of the CNTs@Co–Fe–O, tested in a thermostatic water bath from 0 °C to 10 °C, 30 °C and 50 °C in 1.0 M KOH. | ||
To ascertain the effect of mass loadings of Co0.8Fe0.2Ox/CNTs25 wt% on the activity for OER, we investigated the change of current density with the variation of Co0.80Fe0.20Ox/CNTs25 wt% catalysts on GCE. As shown in Fig. 6c, when the catalyst loadings increase from 0.26 to 0.52 mg cm−2, the current density of the Co0.8Fe0.2Ox/CNTs25 wt% improves. When catalyst loadings exceed 0.52 mg cm−2, the current density starts decreasing obviously. It may be caused by the mass transport problems: when the Co0.80Fe0.20Ox/CNTs25 wt% layers become relatively thick and the bottom of the catalyst layers cannot be utilized efficiently.20,29,33,34,47 Thus, the optimal catalyst loadings of 0.52 mg cm−2 are chosen. Besides, a series of measurements of the Co0.80Fe0.20Ox/CNTs25 wt% catalysts at different temperature (0 °C, 10 °C, 30 °C and 50 °C) have been taken. As presented in Fig. 6d, the Co0.80Fe0.20Ox/CNTs25 wt% exhibit excellent OER activity at large temperature ranges. This is extremely important for expanded application of catalysts for OER.10,33
The electrochemically active surface areas (S) of Co/CNTs25 wt%, Fe3O4/CNTs25 wt%, and Co0.8Fe0.2Ox/CNTs25 wt% were also investigated and presented in Fig. 7. Due to the positive correlation among electrochemical double-layer capacitance (Cdl), S of the materials, and scan rate (ν), that is Cdl ∝ ν × S, S can be calculated from Cdl of the catalytic surface.34,48 By computing the slope between the current density and the ν, the Cdl of Co0.8Fe0.2Ox/CNTs25 wt% is confirmed to be 2330 μF cm−2, which is obvious larger than monocomponent samples (1120 μF cm−2 for Co/CNTs25 wt% and 1390 μF cm−2 for Fe3O4/CNTs25 wt%). As expected, Co0.8Fe0.2Ox/CNTs25 wt% possess a greater active surface area, which is beneficial to supply more active sites exposed to the electrolyte to catalyze OER than other catalysts in this work.
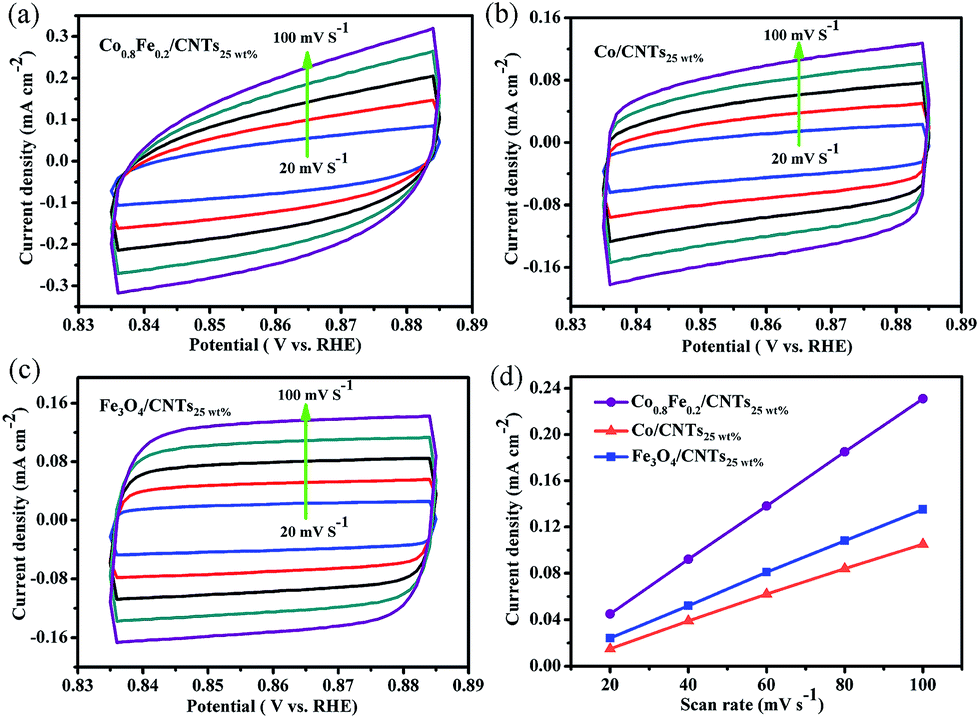 | ||
| Fig. 7 Voltammograms of (a) Co0.8Fe0.2Ox/CNTs25 wt%, (b) Co/CNTs25 wt% and (c) Fe3O4/CNTs25 wt% at various scan rates (20–100 mV s−1) used to (d) estimate the Cdl and relative electrochemically active surface area. | ||
All the conclusions above manifest that Co0.8Fe0.2Ox/CNTs25 wt% exhibited better OER activities than monocomponent Co/CNTs25 wt% and Fe3O4/CNTs25 wt% catalysts, which can be also ascertained from the electrochemical impedance spectroscopy (EIS) measurements that were carried out on Co/CNTs25 wt%, Fe3O4/CNTs25 wt% and Co0.8Fe0.2Ox/CNTs25 wt% at 1.60 V from 105 to 0.1 Hz with a 5 mV AC voltage in O2-saturated 1.0 M KOH. It is worth mentioning that EIS is usually measured to investigate the electrode kinetics, and the diameter of the arc in the Nyquist plots represents the charge transfer resistance (Rct) during the OER process.11,29,49 As shown in Fig. 8a, the Rct for Co0.8Fe0.2Ox/CNTs25 wt% is obviously smaller than other contrast samples, suggesting a decline in reaction resistance. Fig. 8b is the Bode plots for the data in Fig. 8a. Correspondingly, the Bode plots exhibit the details between impedance and frequency. The impedance in the low frequency range is tightly associated to Rct,11,29,50 and it increased in the order Co0.8Fe0.2Ox/CNTs25 wt% < Co/CNTs25 wt% < Fe3O4/CNTs25 wt%, which is in accordance with the Nyquist plots.
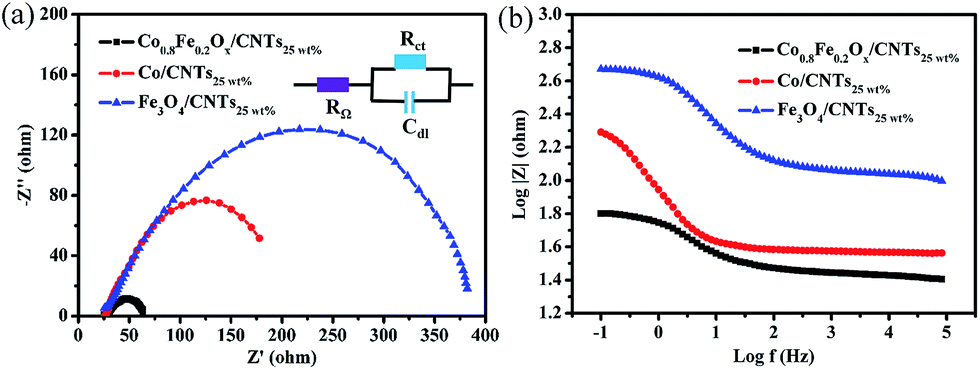 | ||
| Fig. 8 (a) Nyquist diagrams of EIS for Fe3O4/CNTs25 wt%, Co/CNTs25 wt% and Co0.8Fe0.2Ox/CNTs25 wt% in 1.0 M KOH. (b) EIS Bode plots for the catalysts above. | ||
In addition to the activity, durability of catalysts is another very important criteria for OER. Inspiringly, Co0.8Fe0.2Ox/CNTs25 wt% exhibits outstanding durability in 1.0 M KOH. As shown in Fig. 9a, the current density (∼10 mA cm−2, at 1.51 V) can retain for more than 14 h in chronoamperometry (CA) test, while the potential (∼1.58 V, at 10 mA cm−2) can also maintain for more than 14 h in chronopotentiometry (CP) measurement. After we continuously recycle Co0.8Fe0.2Ox/CNTs25 wt% catalysts for 2000 times, the catalysts exhibit the similar LSV performance as before, with insignificant change of the onset potential and current density (Fig. 9b). TEM characterization (inset of Fig. 9b) was also employed and attests the unchanged morphology of the Co0.8Fe0.2Ox/CNTs25 wt% composites after the durability test. To better evaluate the OER activity of the Co0.8Fe0.2Ox/CNTs25 wt% catalysts, physically mixed Co0.8Fe0.2Ox + CNTs25 wt% and the commercial RuO2 catalysts (99.95% metal basis, Alfa Aesar) with the same mass loading (0.52 mg cm−2) were chosen as a reference. As shown in Fig. 9c, the OER performance of Co0.8Fe0.2Ox/CNTs25 wt% catalysts largely exceed the physically mixed Co0.8Fe0.2Ox + CNTs25 wt% catalysts, which suggests that the excellent performance of Co0.8Fe0.2Ox/CNTs25 wt% stems not only from the improved electroconductivity but also from a complicated cooperative effect between Co0.8Fe0.2Ox and CNTs. Moreover, both CA and CP plots (Fig. 9d) demonstrate the low durability of physically mixed Co0.8Fe0.2Ox + CNTs25 wt% within 8 h. Very noticeably, the OER current of Co0.8Fe0.2Ox/CNTs25 wt% largely exceeds that of RuO2 catalysts (Fig. 9e), despite the slightly lower onset potential and η0 of RuO2 catalysts (∼1.38 V and ∼0.25 V, respectively). The Tafel slope value of Co0.8Fe0.2Ox/CNTs25 wt% (∼49 mV dec−1) is lower than commercial RuO2 (∼131 mV dec−1) and exceed that of the previously reported high-performance OER catalysts (Table S1†), suggesting its admirable reaction kinetics. As shown in Fig. 9f, the RuO2 is very unstable in 1.0 M KOH and its potential at 10 mA cm−2 grows quickly during the CP measurement, while the current density at 1.48 V undergos a sharp decline with merely ∼13% maintaining. All the results above indicate the good durability and ultrahigh activity of the Co0.8Fe0.2Ox/CNTs25 wt% catalysts in alkaline solution.
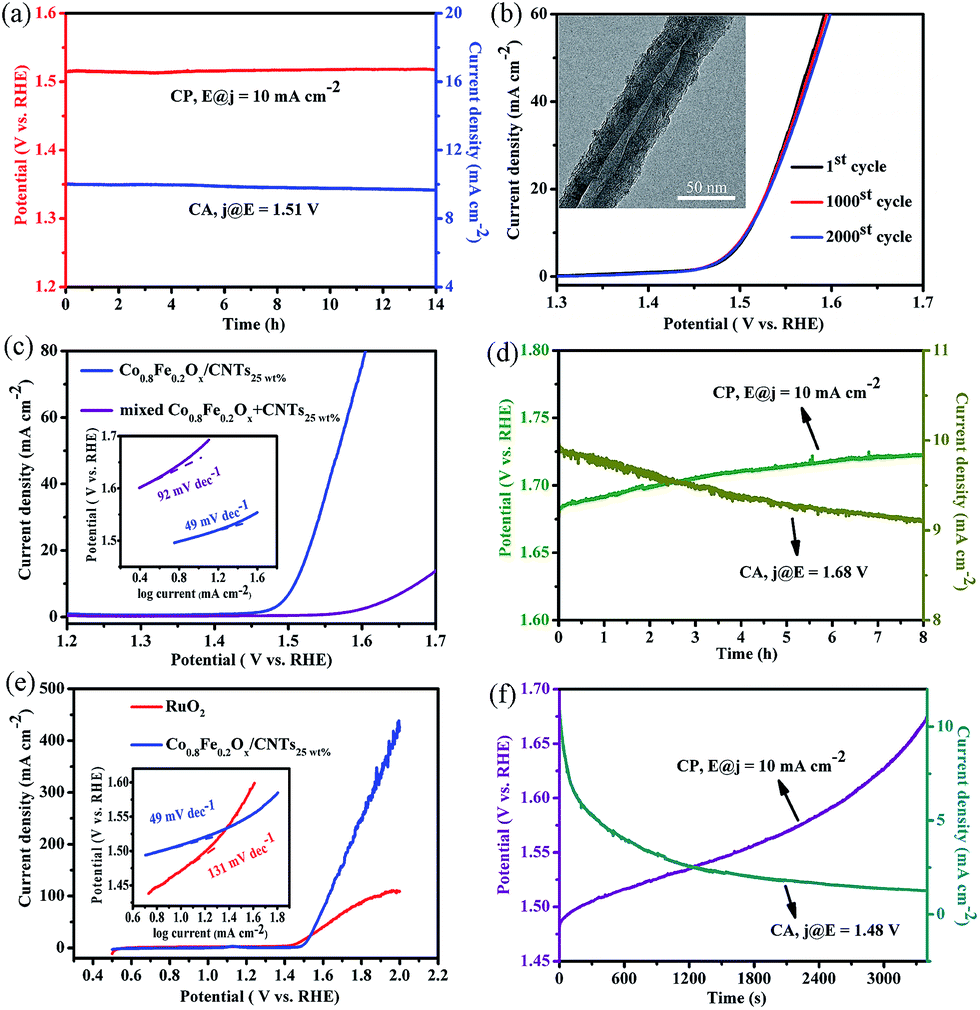 | ||
| Fig. 9 (a) CP and CA plots of the Co0.8Fe0.2Ox/CNTs25 wt%. (b) LSV curves of the Co0.8Fe0.2Ox/CNTs25 wt% before and after 2000 cycles (inset (b): TEM of the Co0.8Fe0.2Ox/CNTs25 wt% after the stability test). (c) LSV curves of Co0.8Fe0.2Ox/CNTs25 wt% and physically mixed Co0.8Fe0.2Ox + CNTs25 wt% in 1.0 M KOH solution; inset in: Tafel plot of the catalysts above. (d) CP and CA plots of the physically mixed Co0.8Fe0.2Ox + CNTs25 wt%. (e) LSV curves of Co0.8Fe0.2Ox/CNTs25 wt% and RuO2 in 1.0 M KOH solution; inset in: Tafel plot of the catalysts above. (f) CP and CA plots of the RuO2. | ||
Conclusions
In summary, we have reported a simple method to synthesize tubular iron-group binary metal nanosheet catalysts Co0.8Fe0.2Ox/CNTs25 wt% through the pyrolysis of Co(acac)2 and Fe(acac)3 in TREG in the presence of CNTs. Via this strategy, carriers CNTs don't requires to introduce other groups for functionalization. The pyrolysis of Co(acac)2 and Fe(acac)3 can result in coaxial ultrathin tubular Co1−yFeyOx nanosheets, without addition of any surfactant or reducing agent. What's more, through discussing their constituent-determined performance and electrochemical impedance spectroscopy for OER, we observe a cooperative effect of Co1−yFeyOx/CNTs composites, which enrich the comprehension of iron-group composites for electrochemical catalysis. The design concept for the iron-group binary metal nanosheets can be spread to prepare other advanced functional materials with improved performance for multifarious applications.Acknowledgements
This research was supported by the National Science Foundation of China (No. 21345003), the Fundamental Research Funds for the Central Universities (Grant no. lzujbky-2016-k08), the Natural Science Foundation of Gansu (145RJZA132), the Key Laboratory of Catalytic Engineering of Gansu Province and Resources Utilization, Gansu Province for financial support.Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- H.-J. Qiu, L. Liu, Y.-P. Mu, H.-J. Zhang and Y. Wang, Nano Res., 2014, 8, 321–339 CrossRef.
- X. Li, Y. Fang, L. Wen, F. Li, G. Yin, W. Chen, X. An, J. Jin and J. Ma, Dalton Trans., 2016, 45, 5575–5582 RSC.
- Y. Feng, H. Zhang, Y. Zhang, X. Li and Y. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 9203–9210 CAS.
- Y. Fang, X. Li, Y. Hu, F. Li, X. Lin, M. Tian, X. An, Y. Fu, J. Jin and J. Ma, J. Power Sources, 2015, 300, 285–293 CrossRef CAS.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920–8930 RSC.
- J. Nai, H. Yin, T. You, L. Zheng, J. Zhang, P. Wang, Z. Jin, Y. Tian, J. Liu, Z. Tang and L. Guo, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- A. Kargar, S. Yavuz, T. K. Kim, C. H. Liu, C. Kuru, C. S. Rustomji, S. Jin and P. R. Bandaru, ACS Appl. Mater. Interfaces, 2015, 7, 17851–17856 CAS.
- W. Bian, Z. Yang, P. Strasser and R. Yang, J. Power Sources, 2014, 250, 196–203 CrossRef CAS.
- P. Li, R. Ma, Y. Zhou, Y. Chen, Z. Zhou, G. Liu, Q. Liu, G. Peng, Z. Liang and J. Wang, J. Mater. Chem. A, 2015, 3, 15598–15606 CAS.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763–17770 CAS.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- X. Ge, Y. Liu, F. W. Goh, T. S. Hor, Y. Zong, P. Xiao, Z. Zhang, S. H. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CAS.
- Y. Zhao, S. Chen, B. Sun, D. Su, X. Huang, H. Liu, Y. Yan, K. Sun and G. Wang, Sci. Rep., 2015, 5, 7629 CrossRef CAS PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, C. Xiao and Y. Xie, Acc. Chem. Res., 2015, 48, 3–12 CrossRef CAS PubMed.
- Y. Liu, C. Xiao, M. Lyu, Y. Lin, W. Cai, P. Huang, W. Tong, Y. Zou and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 11231–11235 CrossRef CAS PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- X. Lu, W.-L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS PubMed.
- X. Li, X.-H. Zhu, Y. Fang, H.-L. Yang, X. Zhou, W. Chen, L. Jiao, H. Huo and R. Li, J. Mater. Chem. A, 2014, 2, 10485–10491 CAS.
- Y. Fang, X. Li, F. Li, X. Lin, M. Tian, X. Long, X. An, Y. Fu, J. Jin and J. Ma, J. Power Sources, 2016, 326, 50–59 CrossRef CAS.
- X. Lu and C. Zhao, J. Mater. Chem. A, 2013, 1, 12053–12059 CAS.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- Z. Sun, Z. Liu, B. Han, Y. Wang, J. Du, Z. Xie and G. Han, Adv. Mater., 2005, 17, 928–932 CrossRef CAS.
- X. Li, Y. Fang, S. Zhao, J. Wu, F. Li, M. Tian, X. Long, J. Jin and J. Ma, J. Mater. Chem. A, 2016 10.1039/c6ta04187f.
- X. Li, Y. Fang, X. Lin, M. Tian, X. An, Y. Fu, R. Li, J. Jin and J. Ma, J. Mater. Chem. A, 2015, 3, 17392–17402 CAS.
- J. Geng, L. Kuai, E. Kan, Q. Wang and B. Geng, ChemSusChem, 2015, 8, 659–664 CrossRef CAS PubMed.
- C. Y. Cao, F. Wei, J. Qu and W. G. Song, Chem.–Eur. J., 2013, 19, 1558–1562 CrossRef CAS PubMed.
- Z. Wang, S. Xiao, Z. Zhu, X. Long, X. Zheng, X. Lu and S. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 4048–4055 CAS.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmekova and T. Asefa, Angew. Chem., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363–5372 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- D. A. Corrigan, R. S. Conel, C. A. Fierro and D. A. Scherson, J. Phys. Chem., 1987, 91, 5009–5011 CrossRef CAS.
- E. B. Castro, C. A. Gervasi and J. R. Vilche, J. Appl. Electrochem., 1998, 28, 835–841 CrossRef CAS.
- S. Palmas, F. Ferrara, A. Vacca, M. Mascia and A. M. Polcaro, Electrochim. Acta, 2007, 53, 400–406 CrossRef CAS.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef CAS PubMed.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2015, 6, 155–161 CrossRef.
- M. Xing, L.-B. Kong, M.-C. Liu, L.-Y. Liu, L. Kang and Y.-C. Luo, J. Mater. Chem. A, 2014, 2, 18435–18443 CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- D.-J. Guo, X.-P. Qiu, W.-T. Zhu and L.-Q. Chen, Appl. Catal., B, 2009, 89, 597–601 CrossRef CAS.
- Z. He and F. Mansfeld, Energy Environ. Sci., 2009, 2, 215–219 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15624j |
| This journal is © The Royal Society of Chemistry 2016 |