
Pressure induced structural transformations of anatase TiO2 nanotubes probed by Raman spectroscopy and synchrotron X-ray diffraction
Abstract
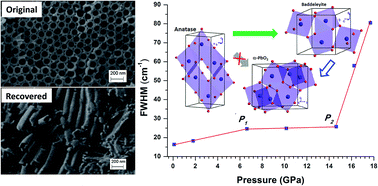
Anatase titanium dioxide (TiO2) nanotubes synthesized via electrochemical anodization were studied under high pressure up to 31 GPa. The structural transformations were characterized by in situ Raman spectroscopy and synchrotron X-ray diffraction. Raman measurements suggest that anatase TiO2 nanotubes transform to an amorphous phase upon compression to 17.7 GPa and remain mostly amorphous upon recovery but with minor α-PbO2 and anatase phases as a mixture. In contrast, direct anatase to baddeleyite phase transition was unambiguously observed at ∼14 GPa in the diffraction measurements. Structural refinement allows the quantitative analysis of the transition sequence and reveals that the recovered phase is mostly crystalline α-PbO2. This discrepancy can be attributed to the special surface and interfacial structure associated with the tube morphology. Moreover, the compression behavior such as the compressibility of both anatase and baddeleyite phases of TiO2 nanotubes was examined in parallel with other nanostructured TiO2 materials. Our analysis shows that morphology plays a more prominent role than size in affecting the high pressure behaviors of 1D TiO2 nanomaterials compared to nanoparticles, and that the interplay of multiple factors such as morphology, size, interfacial structures, as well as lattice defects can substantially influence the phase stability and thus the transformation sequence.
 Please wait while we load your content...
Please wait while we load your content...

