
Different Co-based MOFs templated synthesis of Co3O4 nanoparticles to degrade RhB by activation of oxone†
Jia-yi Pua,
Jin-quan Wan*abc,
Yan Wangac and
Yong-wen Maabc
aCollege of Environment and Energy, South China University of Technology, Guangzhou 510006, China. E-mail: ppjqwan@scut.edu.cn
bState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, China
cThe Key Laboratory of Pollution Control and Ecosystem Restoration in Industry Clusters, Ministry of Education, South China University of Technology, Guangzhou 510006, China
First published on 30th August 2016
Abstract
In this study, Co-based MOF with a 2D structure, 3D microrod structure and 3D nano-dodecahedral structure was synthesized by the coordination of cobalt ions and different organic ligands. Then, Co3O4, which was derived from these Co-based MOFs, was used to activate oxone for RhB degradation. It was found that the morphology and structure of the templates can significantly influence the formation of the nano Co3O4 products, which has an indirect effect on the catalytic ability of the resulting Co3O4 for oxone to degrade RhB. Among the products, nano-sphere Co3O4, which was derived from a template with a 3D nano-dodecahedral structure (ZIF-67) can effectively activate oxone to degrade RhB. Under neutral conditions, the rate constant of the degradation of RhB (0.1 mM) was 0.0509 min−1 with 50 mg L−1 catalyst and 1.0 mM oxone based on first-order kinetics. In addition, the influence of a series of parameter factors (pH, the dosage of catalyst and oxone) on the catalyst was also studied. Low cobalt ion dissolution (0.04 mg L−1 at pH = 3, <0.01 mg L−1 at pH = 7) and good recyclability (degradation rate of RhB after 5 cycles: close to 96%, 89% at pH = 7, pH = 3) further proved that the as-prepared Co3O4 can be used as a promising catalyst.
1. Introduction
In recent years, with the increasingly serious problems of water pollution, sulfate radicals-based advanced oxidation processes (SR-AOPs) has received increasing attention because of their potential capability in the treatment of difficult biological degradation pollutants in water.1 SO4˙− can be produced by the activation of two types of oxidants, peroxymonosulfate (PMS, HSO5−) and persulfate (PS, S2O82−).2 Compared to persulfate, peroxymonosulfate has the following characteristics: (1) in the redox system, PMS can be used as an oxidant and reducing agent at the same time, whereas PS can only be used as an oxidant;3 (2) compared to PS, PMS is activated more easily by transition metals, particularly under neutral conditions;3 (3) unlike PS, PMS can be activated by inorganic anions (e.g., CO32−, HCO3−, HPO42−, and Cl−) in a solution.4 These characteristics endow PMS with prospects in the treatment of actual water pollution. Oxone is the commercial name of potassium peroxymonosulfate (KHSO5·0.5KHSO4·0.5K2SO4).2 Among the various activation systems,5 heterogeneous transition metal/PMS system has been widely studied because it has the characteristics of a simple system, energy saving, less environmental pollution, and convenient catalyst recovery. Transition metal oxide nanoparticles (e.g., Co3O4,6,7 MnO2 (ref. 8) and CuO9) were confirmed to be effective heterogeneous catalysts for PMS activation. However, the performance of nano-metal oxides as a catalyst mostly depends on the morphology, surface areas, particle size, dispersibility, and even structure.7,8,10,11 In addition, fruitful research has been done on Co/PMS systems for organic degradation in waste water treatment.2 To date, researchers have considered the mechanism of the Co/PMS system as the following equations (eqn (1) and (2)).6,7,12,13| Co2+ + HSO5− → Co3+ + SO4˙− + OH− | (1) |
| Co3+ + HSO5− → Co2+ + SO5˙− + H+ | (2) |
It can be noted that the conversion of Co3+ to Co2+ is thermodynamically feasible in this activation reaction, which makes the Co/PMS system very efficient.
Metal organic frameworks (MOFs), which consist of metal ion precursors and organic coordination ligand at the molecular level, have attracted increasing attention owing to its diverse superior properties, such as porous property, high surface area, and diverse structural topology.14 These properties make MOFs attractive in the fields of gas storage and separation, optical devices, sensors, drug delivery, and heterogeneous catalysis.15–19 In addition, the derivative study of using MOFs as templates or precursors to synthesis advanced functional materials, such as metal oxide nano-materials and nano porous carbon materials, has been increasingly popular.12,20–23
Compared to other methods,24–26 the synthesis of metal oxides using MOFs has obvious and unique advantages. First, the preparation method is simple. The preparation of metal oxide nanoparticles using MOF as precursors or templates can only be performed by heat treatment. Second, the resulting metal oxides usually have a porous structure and high surface area. On the one hand, as a template, MOF itself has these characteristics.14 On the other hand, due to the release of small molecules in the process of heat treatment, the resulting products have a porous structure with a high specific surface area.27 Third, this synthesis method is highly controllable. Previous studies have shown that the structure, morphology and particle size of one type of metal-based MOFs may have remarkable distinction due to the differences in the types of organic ligands, ratio, synthesis conditions and other factors. MOF derived metal oxides usually display similar morphology to their templates. Therefore, partial characteristics of the metal oxides can be achieved by pre-controlling the MOF templates.28 For instance, Wu et al. synthesized porous hollow dodecahedra Co3O4 with a high specific surface area (54.5 m2 g−1) and a particle size of about 1 μm for lithium batteries by two-step thermal treatment of ZIF-67 ([Co(MIM)2]n, MIM = 2-methylimidazolate);29 moreover, on using ZIF-67 as the template, Zhang et al. obtained porous nano-dodecahedra Co3O4 with a particle size about 100 nm and a high specific surface area (62.5 m2 g−1) by controlling the size of the template.30 Zhang et al. synthesized Co-based MOF with 1,3,5-benzenetricarboxylate as the organic ligand and used it as template to prepare mesoporous Co3O4 microrod.31 In addition, there are also reports about MOF derived Co3O4 with parallelepipeds,32 flower-like Co3O4 structures,33 and 3D hierarchical urchin-like Co3O4 microspheres.34 However, there is no study on the relationship between the MOF templates and the PMS activation effect.
In this study, we synthesized three types of Co-MOF with the organic ligands of 1,4-benzenedicarboxylic acid (H2BDC), 1,3,5-benzenetricarboxylate (H3BTC) and 2-methylimidazole (2-MIM), which have a 2D structure, 3D micro-rod structure and 3D nano-dodecahedral structure, respectively. Furthermore, Co3O4 nanoparticles were prepared by a two-step heat treatment with these three types of Co-MOF as templates. The resulting products were characterized by X-ray diffraction (XRD), FT-IR spectroscopy, and scanning electronic microscopy (SEM). The experimental results of RhB degradation by oxone showed that the catalytic performance of the as-prepared Co3O4 was affected significantly by its template. In addition, several key parameters in the evaluation of a kinetic study (such as pH value, catalyst and oxone dosage) were also studied. Finally, the stability and recyclability of MOF-derived Co3O4 nanoparticles were tested.
2. Materials and methods
2.1 Materials
The chemicals and reagents in this study are all commercially available and used as received without any further purification. Dye rhodamine B, cobalt nitrate hexahydrate (Co(NO3)2·6H2O), methanol, N,N-dimethylformamide (DMF), and triethylamine were supplied by Kermel Chemical Reagent Co., Ltd (Tianjin, China). 1,4-Benzenedicarboxylic acid (H2BDC), 1,3,5-benzenetricarboxylate (H3BTC), 2-methylimidazolate (2-MIM) and oxone (KHSO5·0.5KHSO4·0.5K2SO4, KHSO5 ≥47%) were supplied by Aladdin Chemistry Co., Ltd (Shanghai, China). Deionized water was purified by a Millipore Milli-Q system.2.2 Synthesis of MOF templates
For 3D Co-MOF with H3BTC as a ligand, its synthesis process is similar to a previously reported method.31 Briefly, Co(NO3)2·6H2O (10 mmol) was dissolved in 50 mL of H2O and stirred magnetically for 20 min. Then, H3BTC (10 mmol) and triethylamine (4 mL) were added to the solution. After stirring for about 30 min, the pink turbid solution was transferred to a 100 mL Teflon-lined stainless steel autoclave and heated to 190 °C for 24 h. The resulting pink product was collected by filtration, washed several times with distilled water and dried in a vacuum oven overnight at 60 °C. The synthetic methods of 2D MOF with H2BDC as the ligand and 3D Co-MOF with 2-MIM as the ligand (ZIF-67) were prepared using the reported procedures.35,362.3 Synthesis of Co3O4 nanoparticles
To maintain the characteristics of the templates, a two-step heat treatment was used in this study.29 First, the as-prepared MOF template was placed into the tubular furnace and carbonized at 650 °C for 5 h under a nitrogen atmosphere to obtain the black powder. The heating rate and gas flow rate were 10 °C min−1 and 50–80 cm3 min−1, respectively. Second, the black powder was placed into a muffle furnace and calcined in air from ambient temperature to 550 °C at a heating ramp of 10 °C min−1 and maintained for 1 h. In order to distinguish easily, the final products derived from the Co-based MOF templates with H2BDC, H3BTC and 2-MIM as the organic ligand were recorded as Co3O4(A), Co3O4(B) and Co3O4(C), respectively.2.4 Characterization
The XRD patterns were obtained on a D8 Advance X-ray diffraction (Bruker-AXS, Karlsruhe, Germany) with a Cu Kα source (λ = 0.15418 nm). Fourier transform infrared spectra (FT-IR) were obtained on a Nicolet Magna 550 FT-IR spectrometer with a KBr disk containing the powder sample. The morphologies and structures of the samples were characterized by thermal scanning electron microscopy (ZEISS, EVO 18, Germany). The specific surface areas were measured using a surface area analyzer (ASAP 2020, Micromeritics).2.5 Catalytic activity measurements
To study the degradation ability of the as-prepared Co3O4/PMS system for RhB, a certain dosage of oxone was added to 200 mL RhB solution (0.1 mM). After that, the solution was adjusted to the desired pH with NaOH (0.1 M) and HCl (0.1 M) solution. A given dosage of catalyst was then added into the solution. To begin the reaction, the flask was shaken at 180 rpm in a rotary shaker (ZHWY-20102C, Shanghai, China) at room temperature. At given time intervals, a 1 mL solution sample was taken out and mixed with 1 mL methanol to quench the residual sulfate radicals immediately. The concentration of the residual RhB was determined by UV-Vis spectrophotometry (Tianmei, UV2310-II) at the maximum wavelength (554 nm). In the recyclability test, the catalysts were collected by centrifugal separation and washed thoroughly with deionized water after each run, then dried at 60 °C for 8 h. In addition, the concentration of leaching Co ion was also determined by atomic absorption spectroscopy (AAS, AA800, PerkinElmer).3. Results and discussion
3.1 Characterization
The crystallographic structure and phase purity of the as-prepared Co3O4 products were examined by X-ray diffraction (XRD) measurement, and the results are shown in Fig. 1. All the diffraction peaks can be indexed to a pure cubic spinel-type Co3O4 (JCPDS card no. 42-1467, space group: Fd3m, lattice constant a = 8.084 Å). No other peaks from residues or contaminants were present, indicating that the products are of high purity.29 In addition, the particles size of Co3O4(A), Co3O4(B) and Co3O4(C) were calculated to be 47.2, 55.4 and 52.4 nm using the Scherrer's equation through the major (222) diffraction peak, which can be further proved by the SEM images (Fig. 3). | ||
| Fig. 1 XRD patterns of the as-prepared Co3O4 catalysts. | ||
As shown in Fig. 2, the FT-IR spectra further confirm that all the three templates were transformed completely to pure Co3O4 without impurities after heat treatment. Only two significant absorption bands could be observed at about 570 and 661 cm−1, which can be attributed to the stretching vibrations of the Co–O bond.37
 | ||
| Fig. 2 FT-IR spectra of the as-prepared Co3O4 catalysts. | ||
The morphology and structure of the Co-based MOF templates and the as-prepared Co3O4 were characterized by scanning electron microscopy. Earlier studies showed that Co-BDC MOF consisted of Co–O–C units, in which the H2BDC links were coordinated with the cobalt centers to form a 2D-polymeric chain.35 As shown in Fig. S1(a) and (b) (see ESI†), Co-BDC presents irregular lamellar morphology. From Fig. S1(c) and (d),† it can be clearly seen that the Co-based MOF, which was synthesized by H3BTC as the organic ligand, consists mainly of quadrangular microrods with a relatively smooth surface. In contrast (Fig. S1(e) and (f)†), another precursor Co-based zeolitic imidazolate framework (ZIF-67) has regular dodecahedral morphology, and the particles size are distributed evenly in 80–100 nm, which is consistent with a previous report.36
Fig. 3 shows the different magnified SEM images of the Co3O4 products. The panoramic view on low-magnified SEM images showed that the obtained Co3O4 products are dissimilar in morphology, which is caused by the difference between the parent MOFs. From the SEM images of Co3O4(A) (Fig. 3(a) and (b)), it can be found that after calcination treatment, the Co3O4 production which derived from Co-MOF template with two dimensional structure agglomerated into irregular blocks. By observing a section of the block, we can observe that it was assembled from two-dimensional sheets. However, when the three-dimensional MOFs were used as templates, the obtained Co3O4 basically maintained the original shape. As shown in Fig. 3(e) and (f), Co3O4(B) successfully retains the microrod morphology after calcination treatment.38 Compared to the template, the surface of Co3O4(B) becomes rough and cracks appear, resulting from the decomposition of the organic species.20 In addition, it can be clearly observed that some of the particles are collapsed, which may be because the calcination temperature is so high that outward gas is released too quickly.29 For Co3O4(C), Fig. 3(i) and (j) show that the product is distributed uniformly, without obvious agglomeration. Fig. 3(k) and (l) demonstrate that the Co3O4(C) particles preserve well the nanometer size and the morphology of ZIF-67. The size range of Co3O4(C) is about 30 to 50 nm, which has been reduced compared to its template. Furthermore, the detailed morphology and structure of the products are supported by the high-magnification images. Fig. 3(c), (d), (g) and (h) show that Co3O4(A) and Co3O4(B) are composed of numerous nanometer-sized particles, which is consistent with the previous XRD results. These nanoparticles can further aggregate to form a porous structure.33 On the one hand, in the crystal structure of metal organic frameworks, metal atoms and organic ligands are arranged regularly, which reduces the distance between metal atoms greatly to form nanoparticles. On the other hand, metal oxidation maintains the original main morphology after calcination due to the limitation of the organic links.39
 | ||
| Fig. 3 SEM images of the as-prepared Co3O4 catalysts at different magnifications: Co3O4(A) (a–d), Co3O4(B) (e–h) and Co3O4(C) (i–l). | ||
To further investigate the specific surface area and the porous nature of the as-obtained Co3O4 products. Brunauer–Emmett–Teller (BET) gas-sorption measurements were performed. Fig. S2 (see ESI†) shows the N2 adsorption–desorption isotherms and pore size distribution of Co3O4(A), Co3O4(B) and Co3O4(C). The detailed results are shown in Table 1. Co3O4(C) presents the highest surface area (14.3 m2 g−1), and Co3O4(B) presents the highest pore volume (0.027 cm3 g−1) and pore width (30.31 nm). Remarkably, the surface area of the as-prepared Co3O4 products is smaller than that previously reported, which should be due to the higher heat treatment temperature.29
| Catalyst | Surface area (SBET, m2 g−1) | Pore volume (cm3 g−1) | Pore width (nm) | Rate constant (min−1) |
|---|---|---|---|---|
| Co3O4(A) | 7.70 | 0.025 | 14.82 | 1.179 × 10−2 |
| Co3O4(B) | 8.95 | 0.027 | 30.31 | 1.778 × 10−2 |
| Co3O4(C) | 14.3 | 0.022 | 6.17 | 5.085 × 10−2 |
3.2 Effect of template on RhB degradation
The oxone-activating ability of Co3O4, which was derived from different Co-based MOFs, was investigated by degrading RhB under various reaction conditions. From Fig. 4, it can be seen that when only the catalyst was added to the solution, no noticeable adsorption of RhB was observed. In the presence of oxone without a catalyst, the removal rate of RhB was about 50% after 120 min, suggesting that oxone itself could partly degrade RhB. However, the degradation of RhB was improved at different degrees by adding the as-prepared Co3O4 catalysts into the system. Co3O4(C)/oxone system exhibited the highest activity for RhB oxidative degradation, nearly 100% of RhB was degraded within 90 min. In contrast, the catalytic activity of Co3O4(A) and Co3O4(B) was much lower, which achieved 76.6% and 88.7% RhB removal in 120 min, respectively. Therefore, the catalytic activity followed the order: Co3O4(C) > Co3O4(B) > Co3O4(A). In order to quantitatively estimate the kinetic rate of RhB removal, a first-order kinetic model was used as follows (eqn (3)):8,12,40,41
 | (3) |
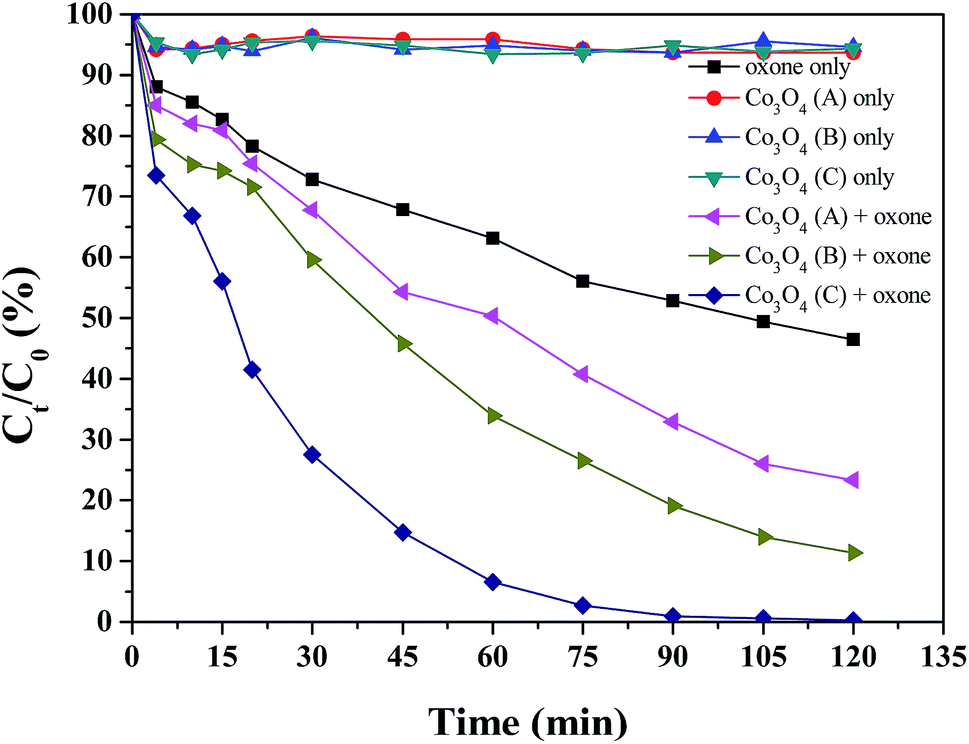 | ||
| Fig. 4 Degradation and adsorption of RhB with Co3O4 derived from different Co-based MOFs. Reaction conditions: RhB = 0.1 mM, catalyst = 50 mg L−1, oxone = 1.0 mM, initial pH = 7.03 and T = 25 °C. | ||
Previous research has shown that the shape, particle size, dispersibility, specific surface area, and even dimensions of the catalyst affect the ability of heterogeneous activated oxone. All of these three as-prepared Co3O4 catalysts are composed of nano-particles, but the difference in their MOF templates gives them a very different morphology by agglomeration, which may affect the number of active sites, resulting in a difference in catalytic ability.
As previously described, the good dispersion, nanometer size and relatively large surface area give Co3O4(C) more active sites for the activation of oxone to generate SO4˙−, which leads to the effective removal of RhB.10,11,42 By comparing Co3O4(A) and Co3O4(B), although Co3O4(B) was reunited into larger micro-particle, the higher specific surface area and pore width give it better catalytic ability.
3.3 Effect of pH on RhB degradation
The influence of the initial solution pH on the heterogeneous as-prepared Co3O4/oxone process was examined by comparing the efficiency and rate constants (k) of RhB degradation. As shown in Fig. 5, Co3O4(C) shows the best catalytic ability in the pH range of 3–9 and the degradation efficiency remained almost the same (Fig. 5(a)). However, as shown in Fig. 5(b), the rate constants of RhB degradation were influenced significantly by the solution pH. It can be seen that relative to acidic and alkaline conditions, the rate constants of RhB degradation were higher under neutral conditions. At low pH, RhB has a positive charge because it is a cationic dye, which is not favorable to absorb on the surface of Co3O4.43 At the same time, oxone exhibits relatively high stability and it is not conducive to the catalytic reaction.2 At high pH condition, the lower degradation rate may be attributed to self-decomposition of oxone at high pH values, which was previously reported.9 | ||
| Fig. 5 Efficiency (a) and rate constants (k) (b) of RhB degradation with Co3O4 derived from different Co-based MOFs at different initial pH within 120 min. Reaction conditions: RhB = 0.1 mM, catalyst = 50 mg L−1, oxone = 1.0 mM, and T = 25 °C. | ||
3.4 Effect of catalyst and oxone dosage on RhB degradation
To further investigate the influence of the catalyst and oxone dosage on the degradation of RhB, a series of control experiments were carried out. As shown in Fig. 6, the degradation of RhB can be influenced greatly by the catalyst dosage. The increase in catalyst dosage could effectively improve the RhB degradation efficiency. RhB could be degraded completely within 120 min at a 25 mg L−1 catalyst dosage. While the catalyst dosage was increased to 100 mg L−1, nearly 100% RhB degradation was achieved within 60 min. By analyzing the change in the rate constants of RhB degradation, it can be found that when the catalyst dosage was increased from 25 to 100 mg L−1, the corresponding reaction rate constants (k) were 0.031 and 0.070 min−1. This was attributed to the more active sites reacting with oxone to make the generation of SO4˙− faster. However, a further increase in catalyst dosage to 150 mg L−1 would only enhance the k value to 0.077 min−1, which may be due to the limitation of oxone dosage.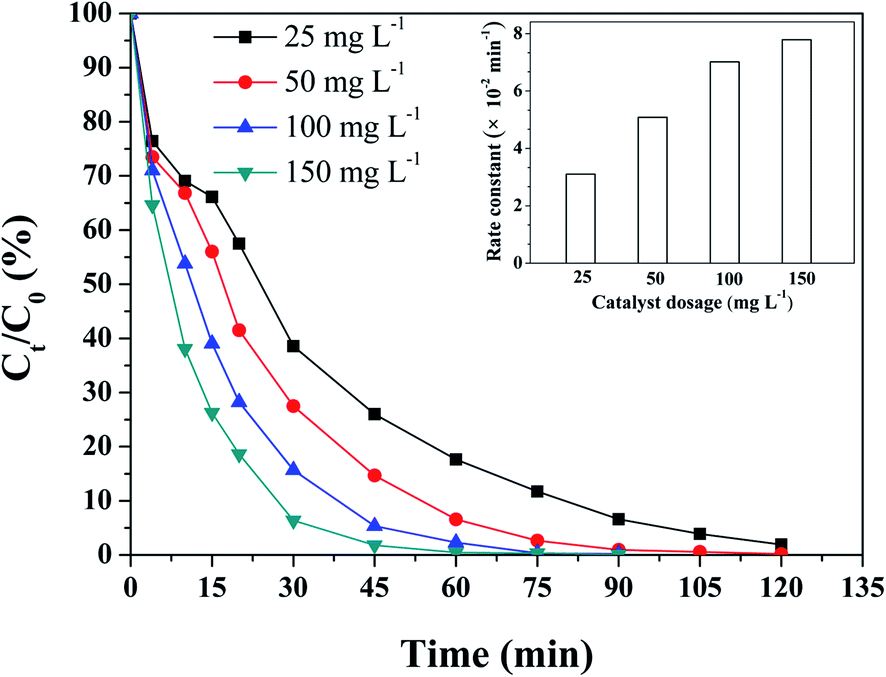 | ||
| Fig. 6 Effect of the catalyst dosage on the degradation of RhB (inset: rate constants at different catalyst dosage). Reaction conditions: RhB = 0.1 mM, oxone = 1.0 mM, initial pH = 7, and T = 25 °C. | ||
Fig. 7 depicts RhB degradation at varying concentrations of oxone. As can be seen, the effect of the oxone dosage was generally similar to the catalyst dosage. A higher concentration of oxone results in a faster degradation rate of RhB. When the dosage of oxone is 0.5 mM, only 94% RhB degradation was achieved after 120 min. While oxone dosage was increased to 1.0, 1.5 and 2.0 mM, RhB can be degraded completely within 90, 60 and 45 min. The k value varies from 0.023 to 0.051, 0.076 and 0.127 min−1 at ambient temperature. Taking the practical application and economic cost into account, 1.0 mM should be the optimal dosage of oxone under this condition.
 | ||
| Fig. 7 Effect of oxone dosage on the degradation of RhB (inset: rate constants at different oxone dosage). Reaction conditions: RhB = 0.1 mM, catalyst = 50 mg L−1, initial pH = 7, and T = 25 °C. | ||
3.5 Stability and recyclability of the catalyst
As a heterogeneous catalyst, the stability and recyclability for the activation of oxone represents important aspects. After the reaction, the leaching of Co ion of the as-prepared three Co3O4 under neutral conditions (pH = 7) and acidic conditions (pH = 3) was tested, and the results are shown in Table 2. It should be noted that the leaching of Co ion, though taking place, remained very low and could be neglected for the catalytic reaction. In addition, the results indicate that the catalyst can be applied to the environment (GB 3838-2002).10| Catalysis | Co2+ leaching under neutral condition (mg L−1) | Co2+ leaching under acidic condition (mg L−1) |
|---|---|---|
| Co3O4(A) | <0.01 | 0.03 |
| Co3O4(B) | <0.01 | 0.03 |
| Co3O4(C) | <0.01 | 0.04 |
Fig. 8 presents the performance of Co3O4(C) after 5 cycles of reuse under neutral and acidic conditions. Co3O4(C) could be used continuously for multiple-cycles and its catalytic activity remained at a high level albeit decreased compared to the first time. The degradation rates of RhB under neutral and acidic conditions were close to 96% and 89%, respectively. In addition, the structure of the catalyst after 5 cycles was basically consistent with it before the reaction by XRD pattern (Fig. S3, see ESI†). These features enable Co3O4(C) to be a durable and effective catalyst for the oxone activation process.
 | ||
| Fig. 8 Recyclability of Co3O4(C) to activate oxone for the degradation of RhB. Reaction conditions: RhB = 0.1 mM, catalyst = 50 mg L−1, oxone = 1.0 mM, reaction time = 90 min and T = 25 °C. | ||
4. Conclusions
In this study, Co3O4 with a different morphology and structure was prepared by two-step heat treatment with different Co-based MOF as templates and then used as a heterogeneous catalyst to activate oxone for RhB degradation in water. The experimental results show that the MOF templates determine the characteristics of the resulting Co3O4 products, which indirectly affect the catalytic ability of as-prepared Co3O4. Co3O4 prepared using ZIF-67 as a template can effectively activate oxone to degrade RhB and also has a very low cobalt ion dissolution rate (0.04 mg L−1 at pH = 3, <0.01 mg L−1 at pH = 7). For the reaction, a higher catalyst and oxone dosage would increase RhB degradation. Kinetic studies showed that RhB degradation followed a first order reaction. The recyclability test also revealed that the as-prepared Co3O4 can be used continuously with stable and effective catalytic activity, suggesting it can be used as a promising catalyst.Acknowledgements
This study was funded by the National Natural Science Foundation of China (No. 31570568), the High-level Personnel Foundation of Guangdong Higher Education Institutions (2013), and the State key laboratory of Pulp and Paper Engineering in China (No. 201535). The authors are thankful to all the anonymous reviewers for their insightful comments and suggestions.Notes and references
- X. M. Lin, Y. W. Ma, Y. Wang, J. Q. Wan and Z. Y. Guan, RSC Adv., 2015, 5, 94694–94701 RSC.
- P. D. Hu and M. C. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38(13), 3705–3712 CrossRef CAS PubMed.
- S. Y. Yang, P. Wang and X. Yang, et al., J. Hazard. Mater., 2010, 179(1–3), 552–558 CrossRef CAS PubMed.
- S. N. Su, W. L. Guo and Y. Q. Leng, J. Hazard. Mater., 2013, 244–245, 736–742 CrossRef CAS PubMed.
- W. L. Guo, S. N. Su, C. L. Yi and Z. M. Ma, Environ. Prog. Sustainable Energy, 2013, 32, 193–197 CrossRef CAS.
- X. Chen, J. Chen, X. Qiao, D. Wang and X. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS.
- Y. Wang, H. Sun, H. M. Ang, M. O. Tade and S. Wang, Appl. Catal., B, 2015, 164, 159–167 CrossRef CAS.
- F. Ji, C. Li and L. Deng, Chem. Eng. J., 2011, 178, 239–243 CrossRef CAS.
- J. Deng, Y. Shao, N. Gao, C. Tan, S. Zhou and X. Hu, J. Hazard. Mater., 2013, 262, 836–844 CrossRef CAS PubMed.
- Y. Yao, Z. Yang, D. Zhang and S. Wang, I&EC Res., 2012, 51(17), 6044–6051 CAS.
- K. Y. Andrew Lin, H. A. Hsuan and R. C. Chen, Chemosphere, 2015, 130, 66–72 CrossRef PubMed.
- P. Shi, X. Wang, X. Zhou, Y. Min, J. Fan and W. Yao, RSC Adv., 2015, 5, 34125–34133 RSC.
- Y. Zhang, J. Wan, Y. Wan and Y. Ma, RSC Adv., 2015, 5, 97589–97597 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112(2), 782–835 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112(2), 869–932 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112(2), 1196–1231 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. Morris and C. Serre, Chem. Rev., 2012, 112(2), 1232–1268 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendor, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112(2), 1105–1125 CrossRef CAS PubMed.
- S. Chen, M. Xue, Y. Li and Y. Pan, et al., Inorg. Chem. Front., 2015, 2, 177–183 RSC.
- X. Sun, Y. Lia, J. Doua, D. Shen and M. Wei, J. Power Sources, 2016, 322, 93–98 CrossRef CAS.
- S. He, Z. Li, J. Wang, P. Wen, J. Gao, L. Ma, Z. Yanga and S. Yang, RSC Adv., 2016, 6, 49478–49486 RSC.
- C. Bao, J. Ma, L. Zhou, Y. Shao, Q. Wu and F. Wang, RSC Adv., 2015, 5, 87616–87625 RSC.
- J. Jiu, Y. Ge, X. Li and L. Nie, Mater. Lett., 2002, 54, 260–263 CrossRef CAS.
- J. Mu, L. Zhang, M. Zhao and Y. Wang, J. Mol. Catal. A: Chem., 2013, 378, 30–37 CrossRef CAS.
- N. N. Binitha, P. V. Suraja, Z. Yaakob, M. R. Resmi and P. P. Silija, J. Sol-Gel Sci. Technol., 2010, 53, 466–469 CrossRef CAS.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 CAS.
- P. Su, S. Liao, F. Rong, F. Wang, J. Chen, C. Li and Q. Yang, J. Mater. Chem. A, 2014, 2, 17408–17414 CAS.
- R. Wu, X. Qian, X. Rui, H. Liu, B. Yadian, K. Zhou, J. Wei, Q. Yan, X. Q. Feng and Y. Long, Small, 2014, 10, 1932–1938 CrossRef CAS PubMed.
- E. Zhang, Y. Xie, S. Ci, J. Jia and Z. Wen, Biosens. Bioelectron., 2016, 81, 46–53 CrossRef CAS PubMed.
- F. Zhang, D. D. Qi and X. G. Zhang, Int. J. Electrochem. Sci., 2016, 11, 189–199 CAS.
- Y. Han, M. Zhao, L. Dong, J. Feng, Y. Wang, D. Li and X. Li, J. Mater. Chem. A, 2015, 3, 22542–22546 CAS.
- L. Zhang, B. Yan, J. Zhang, Y. Liu, A. Yuan and G. Yang, Ceram. Int., 2016, 42, 5160–5170 CrossRef CAS.
- Z. Zhang, L. Li, Q. Xu and B. Cao, RSC Adv., 2015, 5, 61631–61638 RSC.
- F. Zhang, L. Hao, L. Zhang and X. Zhang, Int. J. Electrochem. Sci., 2011, 6, 2943–2954 CAS.
- J. Shao, Z. Wan, H. Liu, H. Zheng, T. Gao, M. Shen, Q. Qu and H. Zheng, J. Mater. Chem. A, 2014, 2, 12194–12200 CAS.
- S. Farhadi, K. Pourzare and S. Sadeghinejad, J. Nanostruct. Chem., 2013, 3, 1–7 Search PubMed.
- F. Ashouri, M. Zare and M. Bagherzadeh, Inorg. Chem. Commun., 2015, 61, 73–76 CrossRef CAS.
- F. Zheng, D. Zhu, X. Shi and Q. Chen, J. Mater. Chem. A, 2015, 3, 2815–2824 CAS.
- K. Y. Andrew Lin and B. C. Chen, Dalton Trans., 2016, 45, 3541–3551 RSC.
- X. Li, Z. Wang, B. Zhang, A. I. Rykov, M. A. Ahmed and J. Wang, Appl. Catal., B, 2016, 181, 788–799 CrossRef CAS.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef CAS.
- J. Zhao, T. Wu, K. Wu, K. Oikawa, H. Hidaka and N. Serpone, Environ. Sci. Technol., 1998, 32, 2394–2400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15590a |
| This journal is © The Royal Society of Chemistry 2016 |