
A palladium–bisoxazoline supported catalyst for selective synthesis of aryl esters and aryl amides via carbonylative coupling reactions†

Abstract
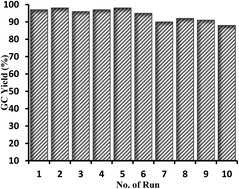
The catalytic synthesis of aryl esters and amides has been successfully achieved in the presence of the efficient palladium–bisoxazoline supported on Merrifield's resin (Pd–BOX-M). The palladium heterogeneous catalyst was prepared and characterized using various spectroscopic techniques. A bisoxazoline ligand having a suitable functional group (BOX-OH) was first synthesized, characterized, chemically supported on Merrifield's resin, and finally coordinated to palladium(II) chloride. The catalytic activity and the recycling ability of the new palladium supported catalyst have been investigated in the alkoxycarbonylation and aminocarbonylation of various aryl iodides using different alkyl and aromatic alcohols and amines as nucleophiles. The palladium heterogeneous catalyst demonstrated excellent catalytic activity and very high recycling ability in the above two carbonylation reactions. The palladium heterogeneous catalysts showed an excellent stability under carbon monoxide and under the experimental conditions.
 Please wait while we load your content...
Please wait while we load your content...

