
Tribromide ion immobilized on magnetic nanoparticle as a new, efficient and reusable nanocatalyst in multicomponent reactions†
Maryam Hajjami* and
Fatemeh Gholamian
Ilam University, Iran. E-mail: mhajjami@yahoo.com; m.hajjami@ilam.ac.ir
First published on 29th August 2016
Abstract
Tetraethyldiethylenetriamine tribromide magnetic nanoparticles (MNPs-TEDETA tribromide) are synthesised and characterized as an efficient, new, metal free and magnetically reusable nanocatalyst for the synthesis of 2,3-dihydroquinazolin-4(1H)-one and polyhydroquinoline derivatives. The synthesized catalyst was characterized using FT-IR, TGA, XRD, EDX, VSM and SEM analyses.
1 Introduction
There are two main kinds of catalysis systems, heterogeneous and homogeneous, and each of them has advantages and drawbacks. For example, the benefits of heterogeneous catalysts are: easy synthesis, easy separation, efficient recycling, easy product purification, and the disadvantages include: insufficient catalytic surface, low activity and leaching. Meanwhile, homogeneous catalysts have benefits such as: high activity and high selectivity, but they have disadvantages such as: laborious product purification, difficulty in recycling and recovery of the catalyst, and deactivation of the catalyst towards the end of the reaction.1–3 Because the catalysts are often expensive, the reuse and separation of them are very important. To overcome these drawbacks, magnetic nanoparticles (MNPs), which are often low cost, have easy and clean separation from the reaction mixture with an external magnet,4–6 unique properties, potential applications in a variety of fields,7 simple synthesis, high surface area, low toxicity and are readily available, are preferred.8Multicomponent reaction (MCR) condensations are a chemical reaction where three or more simple substrates react to give highly selective products that retain the majority of the atoms of the starting materials.9 MCRs are an important subclass of tandem reactions which have advantages such as a convergent nature, milder reaction conditions, simplicity, high atom economy, easy techniques, shorter reaction time, are environmentally friendly, a lower cost and energy conservation.10,11 In the past decade the methodologies of MCRs have emerged as being very efficient ways to access heterocycles.12
Polyhydroquinoline and 2,3-dihydroquinazolinone derivatives are very well-known molecules that include a six membered N-heterocyclic ring. These compounds have been reported to possess a vast range of biological properties and pharmaceutical activities.8,13 2,3-Dihydroquinazolinones are a class of N-heterocycles that have attracted much attention because of their broad spectrum of pharmacological, biological14 and medicinal activities such as being antibacterial, antifertility, antitumor, antifibrilatory, vasodilatory, antifungal, and analgesic.15,16 In addition, quinazolines are oxidized into quinazolin-4(3H)-ones analogs, which are known to be important biologically active heterocyclic compounds.17 2,3-Dihydroquinazolin-4(1H)-one analogues have been synthesized using several catalysts such as: copper(II) chloride/iron(II,III) oxide (CuCl2/Fe3O4)-tetraethyldiethylenetriamine (TEDETA),4 glucosulfonic acid immobilized on Fe3O4 MNPs (GSA@MNPs),14 BiBr3,18 2-morpholinoethanesulfonic acid (MES),19 SiO2–FeCl3,20 boehmite silica sulfuric acid (boehmite-SSA),21 boehmite silica dopamine sulfamic acid (boehmite-Si-DSA).22
Some of these methodologies for the synthesis of 2,3-dihydroquinazolin-4(1H)-one which have been previously reported had expensive reagents, tedious work-ups, high reaction temperatures, harsh reaction conditions and low yields. Thus, development of a versatile and simple procedure is a highly desired goal in the synthesis of 2,3-dihydroquinazolin-4(1H)-ones.16,23 Although some of the analogue nanoparticles prepared by other authors were previously reported,24,25 the main novelty of this research is it is the first report of MNPs-TEDETA tribromide without any transition metals being used as the catalyst for MCRs.
In this work, the aim was to develop an efficient synthetic process, for the synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives using a one-pot, two-component condensation of anthranilamide and aldehydes catalysed by MNPs-TEDETA tribromide in ethanol (EtOH) under reflux conditions and synthesis of polyhydroquinoline by MCRs of aldehyde, dimedone, ethyl acetoacetate (EtOAc), ammonium acetate and MNPs-TEDETA tribromide as catalyst in poly(ethylene glycol) (PEG) at 80 °C.
2 Results and discussion
2.1 Catalyst preparation
In this paper a new and efficient method for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones and polyhydroquinolines in the presence of catalytic amounts of MNPs-TEDETA tribromide is reported. Scheme 1 details the synthesis procedure of the catalyst. Initially the black precipitate of the magnetic nano particles of Fe3O4 have been prepared using a mixture of FeCl3·6H2O and iron(II) chloride tetrahydrate (FeCl2·4H2O) in 30% ammonium hydroxide (NH4OH) under a N2 atmosphere at 80 °C.4,13 Then, Fe3O4 NPs were coated with (3-chloropropyl)trimethoxysilane (CPTMS) solution. In the next step, the mixture of TEDETA and triethylamine (Et3N) was added to the MNPs-CPTMS under reflux with dry toluene. Finally, the TEDETA functionalized MNPs (MNPs-TEDETA) obtained were reacted with hydrogen bromide (HBr) and bromine (Br2) to give MNPs-TEDETA tribromide.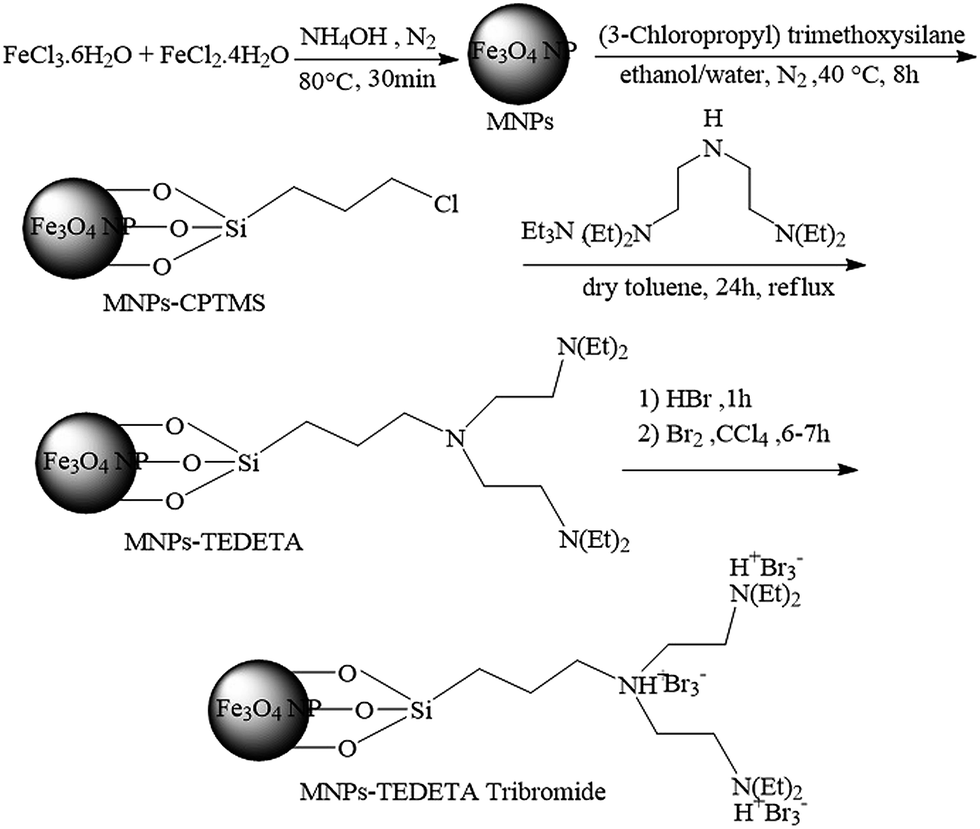 | ||
| Scheme 1 Synthesis of MNPs-TEDETA tribromide. | ||
2.2 Catalyst characterization
All reagents and solvents were purchased from Merck and Aldrich chemical companies. The synthesised catalyst was characterized using X-ray diffraction (XRD, MMA-Mini Materials Analyzer, GBC-Difftech), Fourier transform infrared spectroscopy (FT-IR, Bruker, Germany), scanning electron microscopy (SEM, MIRA3 field emission scanning electron microscope (FESEM), Tescan), energy dispersive X-ray (EDX, MIRA3 FESEM, Tescan), thermogravimetric analysis (TGA, Pyris Diamond differential scanning calorimeter, Perkin-Elmer, UK), and vibrating sample magnetometry (VSM, MDKFD).As seen in Fig. 1, the position of all the peaks in the XRD pattern of MNPs-TEDETA tribromide and the recovered catalyst were in agreement with the standard XRD pattern of Fe3O4. As shown the catalyst and the recovered catalyst were identified from the peak positions at 2θ = 21.33, 35.20, 41.55, 50.65, 63.19, 67.52, 74.53 and 2θ = 21.32, 35.09, 41.45, 50.53, 63.27, 67.81, 74.58, respectively. These peaks with the corresponding reflections of (111), (220), (311), (400), (422), (511) and (440) indicated that the surface modification of the Fe3O4 NPs does not lead to their phase change, even after recovery and they all match well with the data for the standard Fe3O4 sample.26 Also this pattern indicated that the crystalline inverse cubic spinel structures are protected during the functionalization of the MNPs.
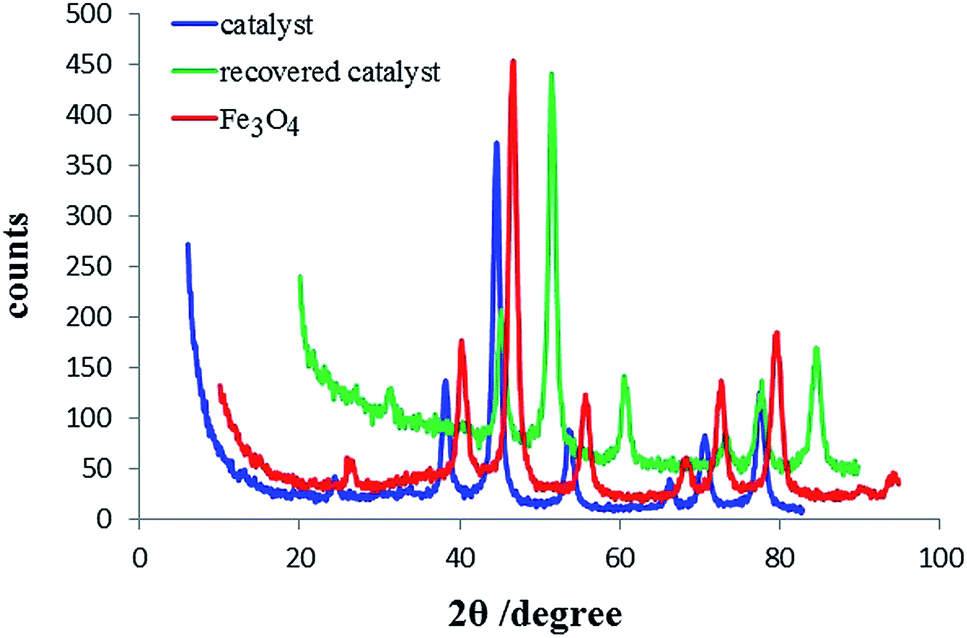 | ||
| Fig. 1 XRD pattern of MNPs-TEDETA tribromide and recovered catalyst. | ||
Fig. 2 shows the FT-IR spectra for the MNPs, MNPs-CPTMS, MNPs-TEDETA, and MNPs-TEDETA tribromide. The IR spectrum of the Fe3O4 alone indicates the bond stretching vibration at 3401 cm−1 belongs to O–H bonds which are attached to the surface iron atoms and a peak appears at 1625 cm−1 which belongs to the stretching vibrational mode of an adsorbed H2O layer. Also the peak that appeared at 579 cm−1 comes from vibrations of Fe–O bonds.14,27
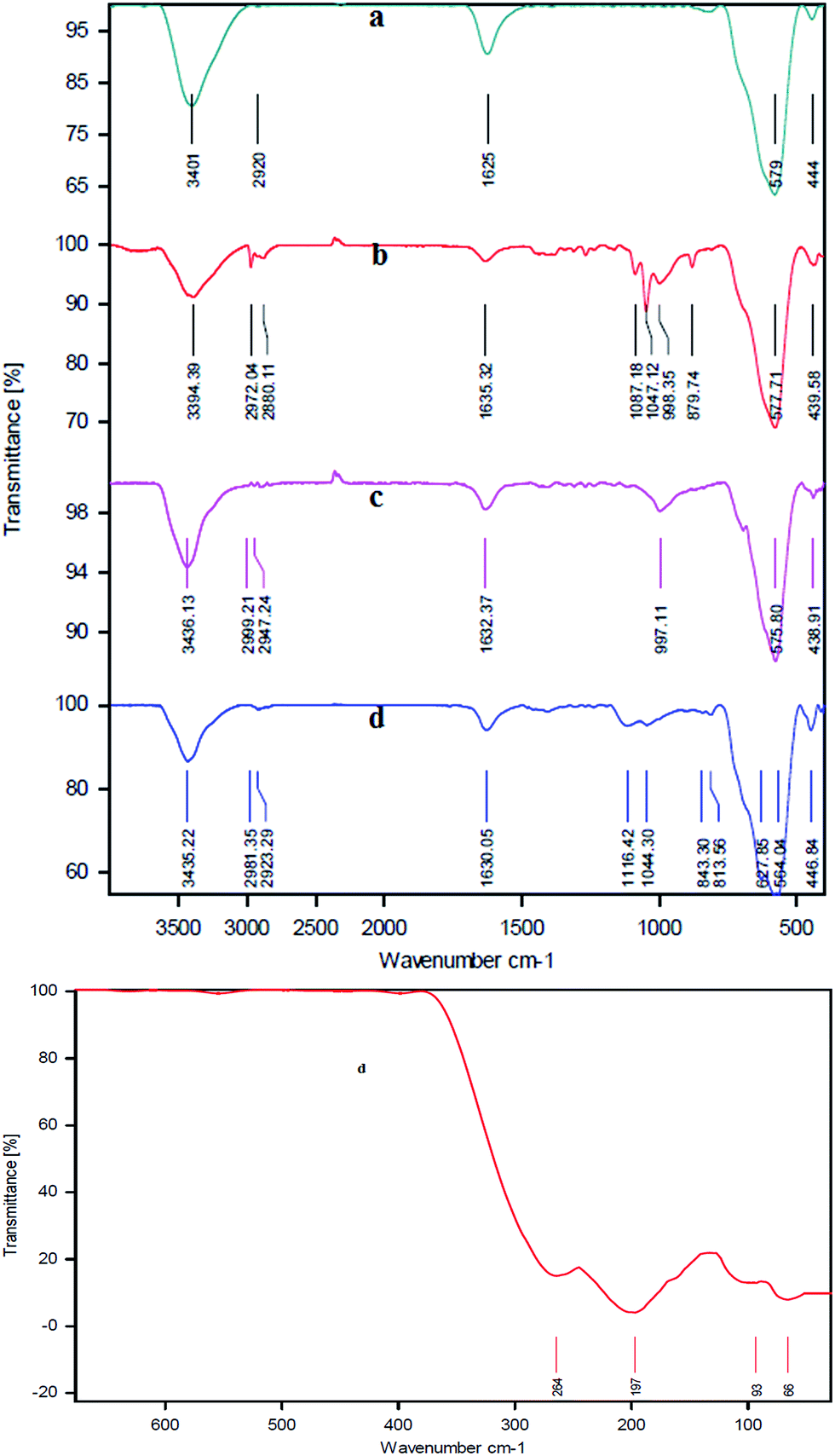 | ||
| Fig. 2 FT-IR spectra for the MNPs (a), MNPs-CPTMS (b), MNPs-TEDETA (c), and MNPs-TEDETA tribromide (d). | ||
The IR spectrum of MNPs-CPTMS [Fig. 2(b)] shows the peak at 577 cm−1 which is assigned to the Fe–O vibration. The band formation between MNPs and the 3-chloropropylsilica group is confirmed by the Fe–O–Si absorption band that appears at near 999 cm−1.28 The peaks at 2880 cm−1 and 2972 cm−1 are attributed to C–H stretching vibrations.27,29
As shown in Fig. 2(c), the presence of bands at 575 cm−1 and 997 cm−1 are assigned to the Fe–O vibration14,27 and Fe–O–Si absorption,28 respectively. The peaks at 2947 cm−1 and 2999 cm−1 regions are attributed to the C–H stretching vibrations.29
The IR spectrum of the MNPs-TEDETA tribromide [Fig. 2(d)] indicates that the bonds at 564 cm−1 and 627 cm−1 are assigned to the vibrations of Fe–O bonds. The presence of a band at 1630 cm−1 belongs to the stretching vibrations of C–N+.24 This IR spectrum also shows the peaks at 1040 cm−1 and 1116 cm−1 which are assigned to the SiO–H and Si–O–Si groups.30 The presence of bands in the 2923 cm−1 and 2981 cm−1 regions are assigned to the C–H stretching vibrations.27,29 For the peak of Br–Br indicated, which is exhibited below 400 cm−1, another FT-IR spectrum was captured in the region below 400 cm−1. As shown in Fig. 2, the peaks at 197 and 264 cm−1 are assigned to Br3.
The SEM image was used to investigate the morphology and size of the catalyst particles. This confirmed that the catalyst was made up of uniform sized particles with an average diameter of less than 10 nm and also that the shape of the MNPs-TEDETA tribromide is spherical (Fig. 3).
 | ||
| Fig. 3 SEM images of the MNPs-TEDETA tribromide. | ||
The EDX spectrum of the catalyst indicates the elements present (Br, C, Fe, N, O, and Si) in the MNPs-TEDETA tribromide. Fig. 4(a) shows the EDX spectrum of the recovered catalyst and Fig. 4(b) shows the EDX spectrum of the recovered catalyst.
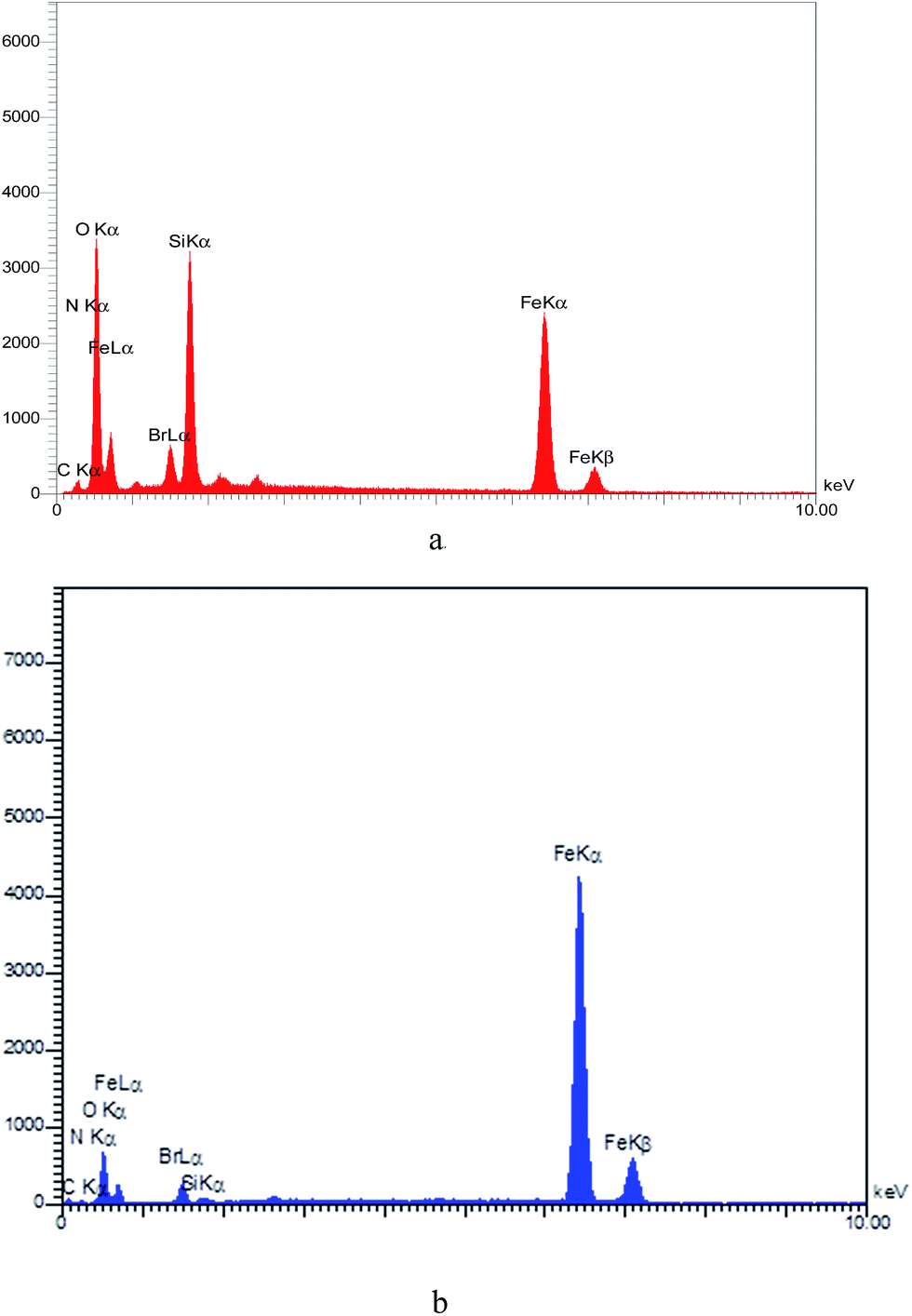 | ||
| Fig. 4 EDX spectrum for MNPs-TEDETA tribromide (a), recovered catalyst (b). | ||
The bond formation between the NPs and the catalyst can be inferred from the TGA results. The TGA curves of the MNPs-TEDETA tribromide and bare Fe3O4 nanoparticles are shown in Fig. 5. The mass loss at temperatures below 200 °C is because of the removal of physically adsorbed surface hydroxyl groups and solvent. The weight loss of 7% for MNPs-TEDETA tribromide between 205 and 636 °C is associated with the thermal decomposition of organic groups grafted to Fe3O4.
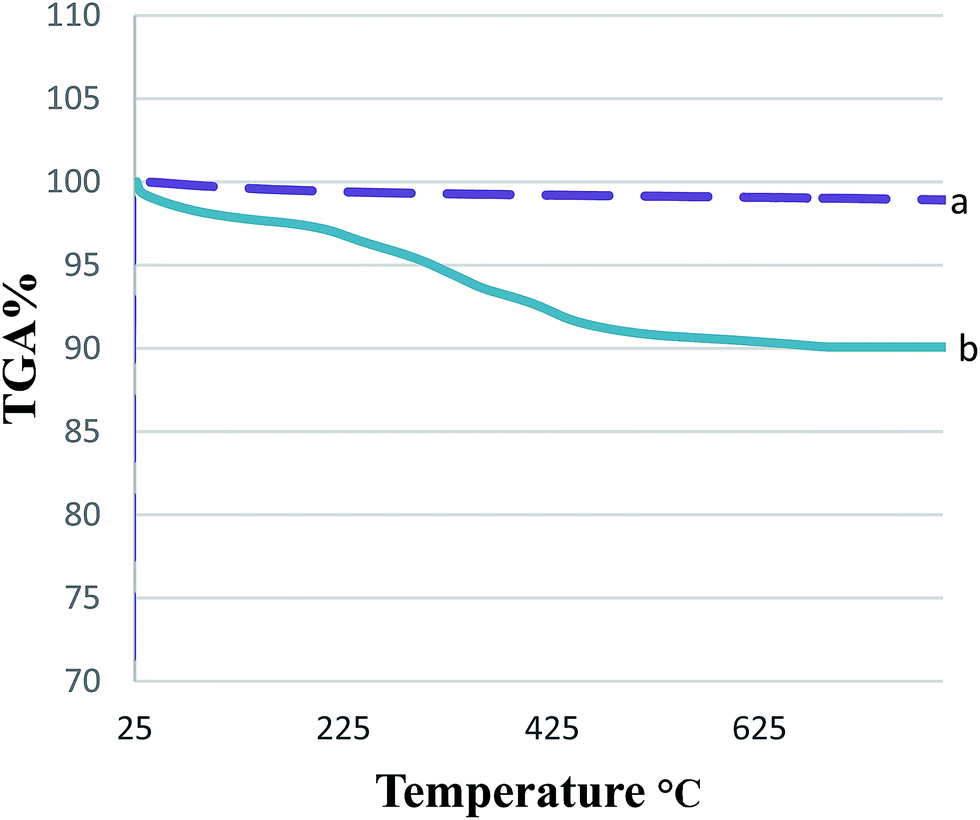 | ||
| Fig. 5 TGA profile of Fe3O4 (a) and MNPs-TEDETA tribromide (b). | ||
VSM analysis is employed for the measurement of the magnetic properties of MNPs and MNPs-TEDETA tribromide. As shown in Fig. 6, the bare MNPs and MNPs-TEDETA tribromide both show excellent paramagnetism but because of the coating of silica and the layer of the attached catalyst, the bare MNPs had a higher magnetic value in comparison with the MNPs-TEDETA tribromide.
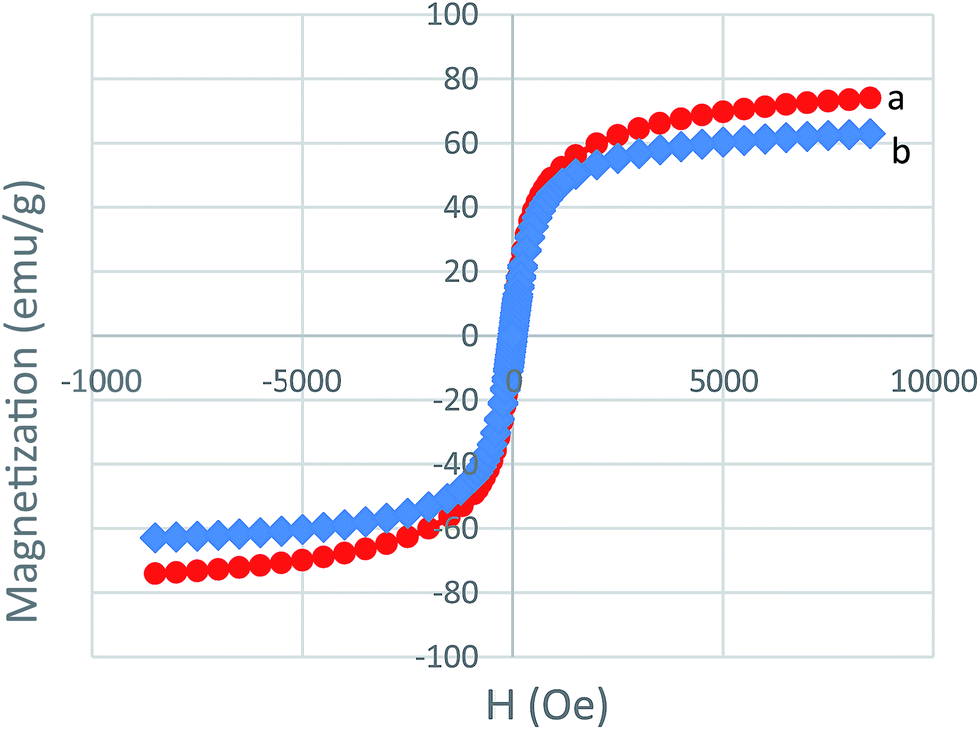 | ||
| Fig. 6 VSM analysis of Fe3O4 (a) and MNPs-TEDETA tribromide (b). | ||
2.3 Catalytic study
After characterizing the MNPs-TEDETA tribromide, its efficiency as nanocatalyst in organic reactions such as synthesis of 2,3-dihydroquinazolin-4(1H)-one and polyhydroquinoline derivatives was investigated. First, to optimize the reaction conditions for the synthesis of 2,3-dihydroquinazolin-4(1H)-one, the condensation of 4-chlorobenzaldehyde (1 mmol) with 2-aminobenzamide and MNPs-TEDETA tribromide as catalyst was used as a model reaction (Scheme 2). The reactions were carried out in the presence of different amounts of catalyst and 2-aminobenzamide and various solvents. The best results were obtained with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.05 for aldehyde:2-aminobenzamide and 0.05 g of MNPs-TEDETA tribromide in EtOH under reflux conditions (Table 1, entry 3).
:1.05 for aldehyde:2-aminobenzamide and 0.05 g of MNPs-TEDETA tribromide in EtOH under reflux conditions (Table 1, entry 3).
 | ||
| Scheme 2 MNPs-TEDETA tribromide catalysis of the one-pot synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives. | ||
| Entry | 2-Aminobenzamide (mmol) | Catalyst (g) | Solvent | Temperature (°C) | Yieldb (%) |
|---|---|---|---|---|---|
| a 80 min.b Isolated yield.c NR = no reaction. | |||||
| 1 | 1.05 | 0.03 | EtOH | 80 | 63 |
| 2 | 1.05 | 0.04 | EtOH | 80 | 69 |
| 3 | 1.05 | 0.05 | EtOH | 80 | 95 |
| 4 | 1.05 | 0 | EtOH | 80 | NRc |
| 5 | 1.05 | 0.06 | EtOH | 80 | 93 |
| 6 | 1.1 | 0.05 | EtOH | 80 | 88 |
| 7 | 1 | 0.05 | EtOH | 80 | 81 |
| 8 | 1.05 | 0.05 | EtOH | 70 | 70 |
| 9 | 1.05 | 0.05 | H2O | 80 | NR |
| 10 | 1.05 | 0.05 | CH2Cl2 | 80 | 37 |
| 11 | 1.05 | 0.05 | EtOAc | 80 | 45 |
| 12 | 1.05 | 0.05 | n-Hex | 80 | 15 |
| 13 | 1.05 | 0.05 | PEG | 80 | Trace |
| 14 | 1.05 | 0.05 | Acetone | 80 | 32 |
In general, the choice of solvent can have a significant effect on the performance of a reaction. Solvents can have an effect on solubility and reaction rates. To determine if this was the case, the synthesis of 2,3-dihydroquinazolin-4(1H)-one using different solvent (polar and non-polar) such as: acetone, CH2Cl2, EtOAc, EtOH, H2O, n-Hex, and PEG was monitored. The green solvent of H2O was applied but no product was obtained (no reaction). Also with PEG there was only a trace yield of product. Although acetone, CH2Cl2, EtOAc, and n-Hex are not safe solvents, they were investigated further. As shown in Table 1, these solvents had low yield (15–45%) and were not suitable for this MCR. Among solvents tested, EtOH was the best solvent because it significantly improved the yield and it is safer than using acetone, CH2Cl2, EtOAc, or n-Hex. Also in several methods reported previously for the synthesis of 2,3-dihydroquinazolin-4(1H)-one, the solvent of reaction was EtOH.14,19,21,31
The reaction of the various benzaldehyde derivatives was then investigated. The 2,3-dihydroquinazolin-4(1H)-one derivatives were obtained in high yields. The results of this study are summarized in Table 2. As shown, a variety of benzaldehydes bearing electron donation and electron withdrawing substituents were successfully employed to prepare the corresponding 2,3-dihydroquinazolin-4(1H)-one derivatives with excellent yields.
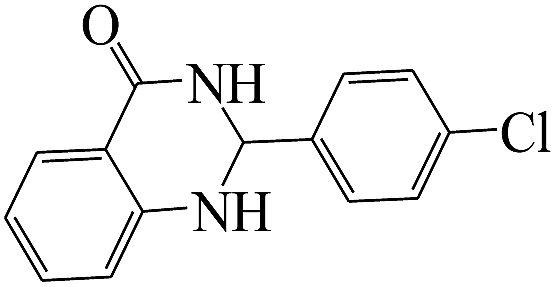
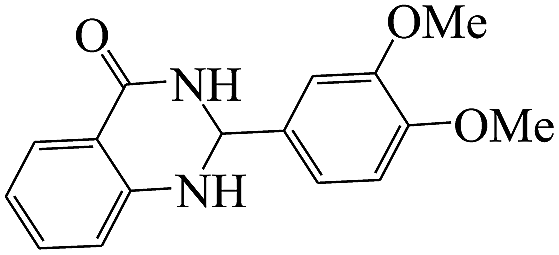
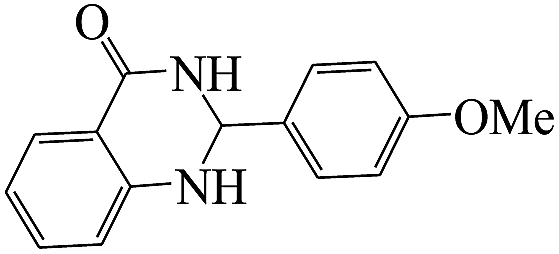
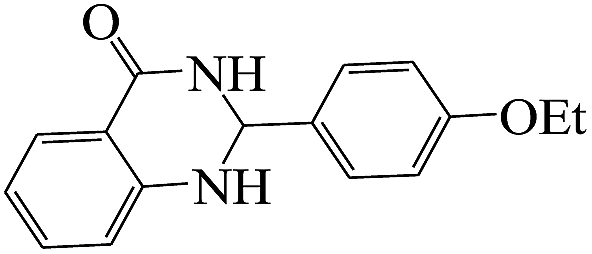
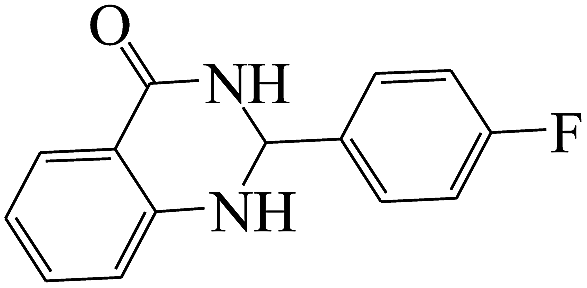
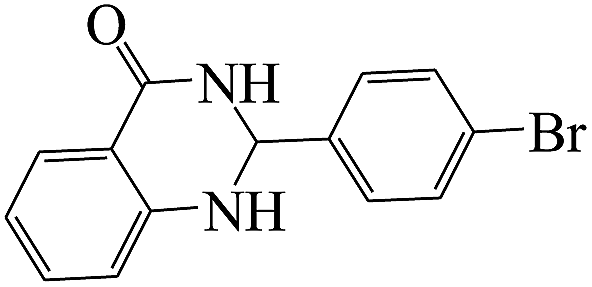
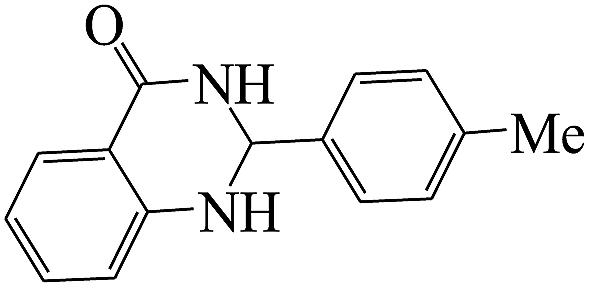
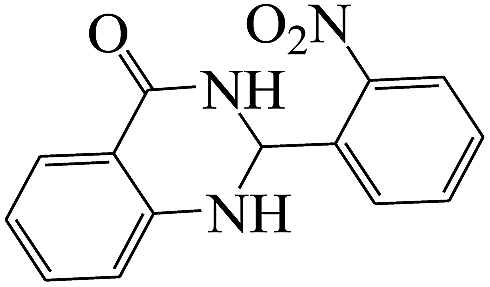
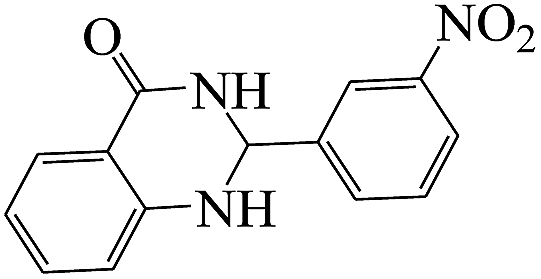
The plausible mechanism for the formation of 2-aryl-2,3-dihydroquinazolinone-4(1H)-one was proposed and is shown in Scheme 3. The carbonyl group in aldehyde was activated by the catalyst. Then the anthranilamide reacted with the activated aldehyde and the intermediate I is formed. After dehydration of intermediate I, the imine intermediate II is generated. Finally the intramolecular cyclization of imine intermediate II produced the final product.32
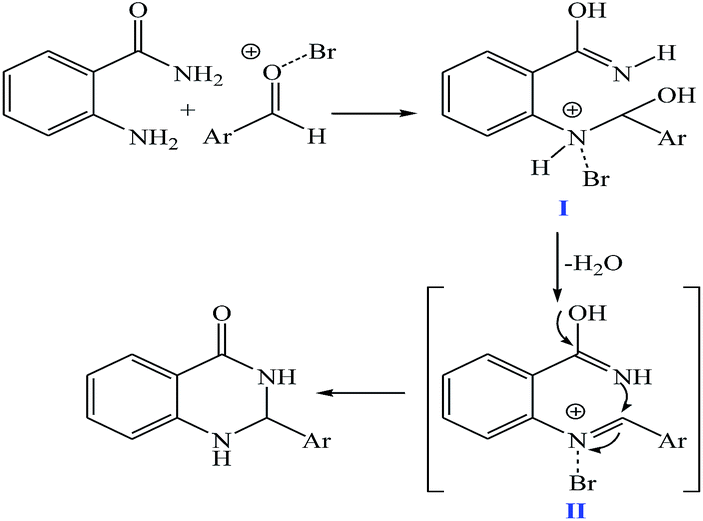 | ||
| Scheme 3 The proposed mechanism of the synthesis of 2-aryl-2,3-dihydroquinazolinone-4(1H)-one. | ||
In the second part of this work, finding a method for the one-pot synthesis of polyhydroquinoline derivatives using MNPs-TEDETA tribromide as a recoverable nanocatalyst in PEG as a green solvent (Scheme 4) was investigated.
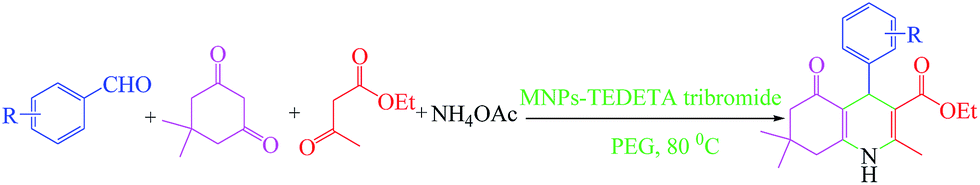 | ||
| Scheme 4 Synthesis of polyhydroquinoline derivatives catalyzed by MNPs-TEDETA tribromide. | ||
To optimize the reaction conditions for the one-pot synthesis of polyhydroquinoline derivatives, 4-chlorobenzaldehyde with dimedone, ammonium acetate and EtOAc was used as a model reaction in the presence of different organic solvents and different amounts of catalyst and ammonium acetate at various temperatures and conditions (Table 3).
| Entry | Catalyst (g) | Ammonium acetate (mmol) | Solvent | Temperature (°C) | Time (min) | Yielda (%) |
|---|---|---|---|---|---|---|
| a Isolated yield. | ||||||
| 1 | 0.04 | 1.2 | PEG | 80 | 300 | 78 |
| 2 | 0.05 | 1.2 | PEG | 80 | 120 | 92 |
| 3 | 0.06 | 1.2 | PEG | 80 | 140 | 93 |
| 4 | 0 | 1.2 | PEG | 80 | 120 | 20 |
| 5 | 0.05 | 1.2 | PEG | 60 | 120 | 55 |
| 6 | 0.05 | 1.2 | PEG | 100 | 120 | 67 |
| 7 | 0.05 | 1.1 | PEG | 80 | 120 | 74 |
| 8 | 0.05 | 1 | PEG | 80 | 120 | 70 |
| 9 | 0.05 | 1.2 | H2O | 80 | 120 | Trace |
| 10 | 0.05 | 1.2 | EtOH | 80 | 120 | 61 |
| 11 | 0.05 | 1.2 | EtOAc | 80 | 120 | 50 |
As shown in entry 2 of Table 3 after the optimization of the reaction conditions, 4-chlorobenzaldehyde (1 mmol), dimedone (1 mmol), EtOAc (1 mmol) and ammonium acetate (1.2 mmol) in the presence of 0.05 g MNPs-TEDETA tribromide in PEG showed better results in terms of the reaction yield and rate.
In this reaction, various benzaldehyde derivatives were employed. As shown in Table 4, the polyhydroquinoline derivatives were obtained in high yields.
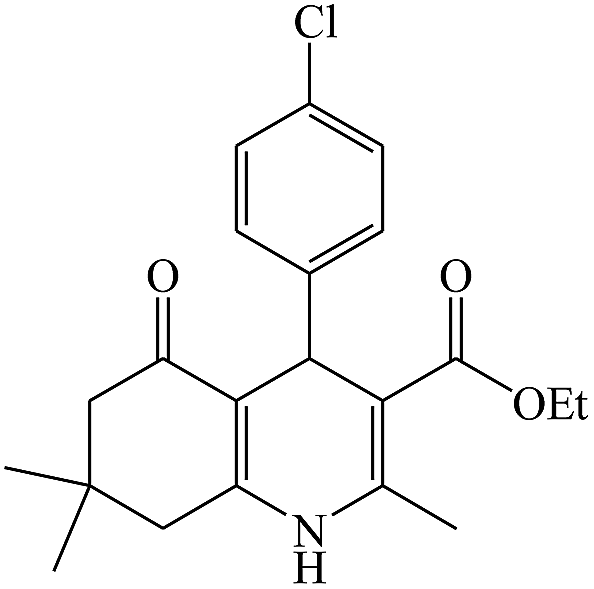
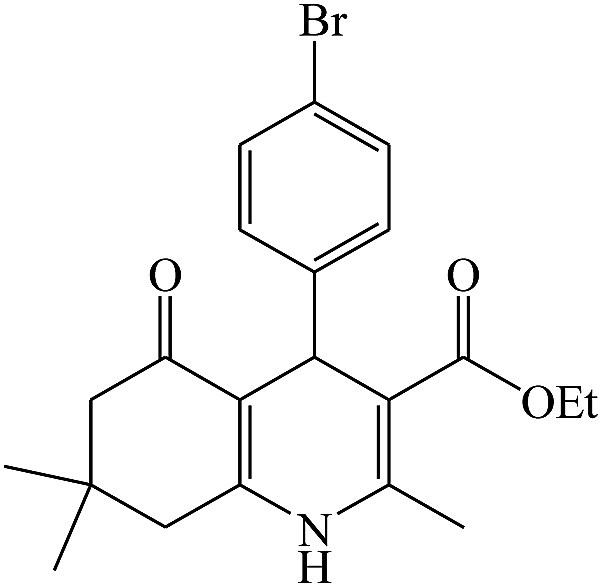
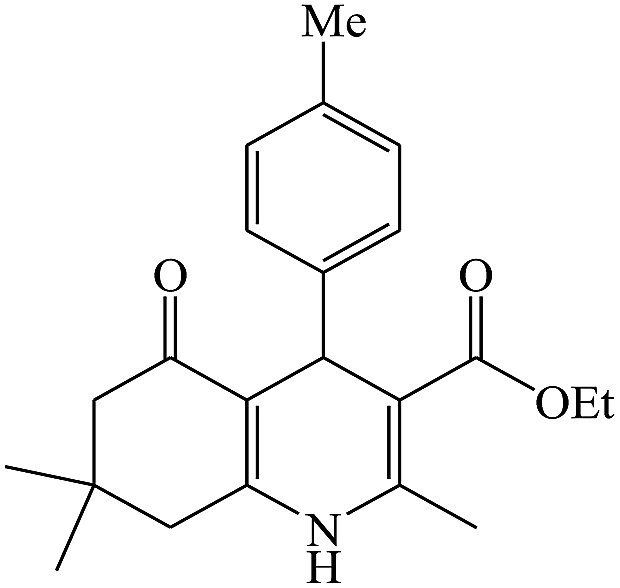
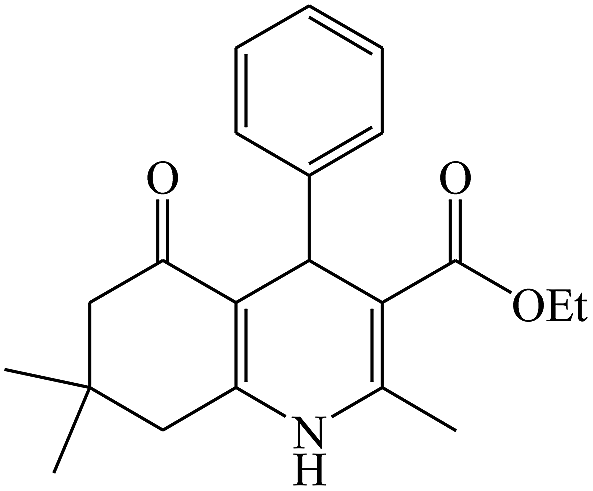
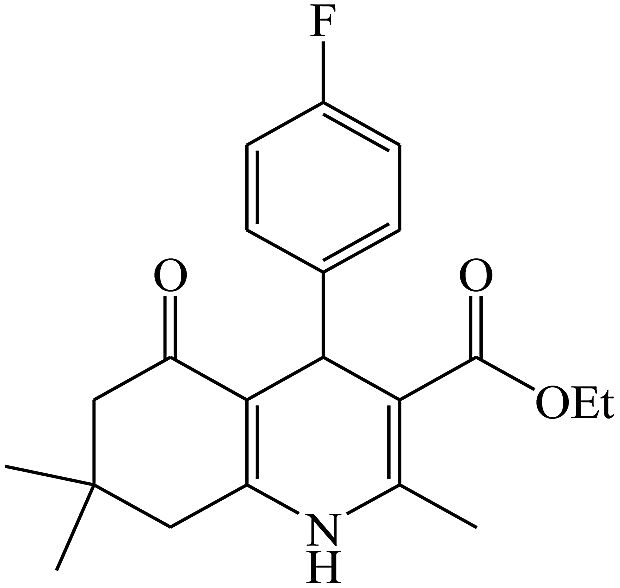
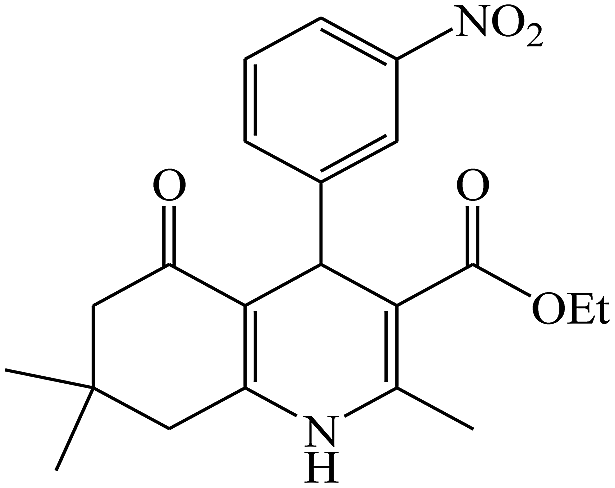
Scheme 5 shows the mechanism of the synthesis of polyhydroquinoline derivatives.14 The role of the catalyst comes in the condensation of aldehydes with the active methylene compounds (Knoevenagel condensation) to give an α,β-unsaturated compound and the Michael-type addition of the intermediates to produce the polyhydroquinoline.
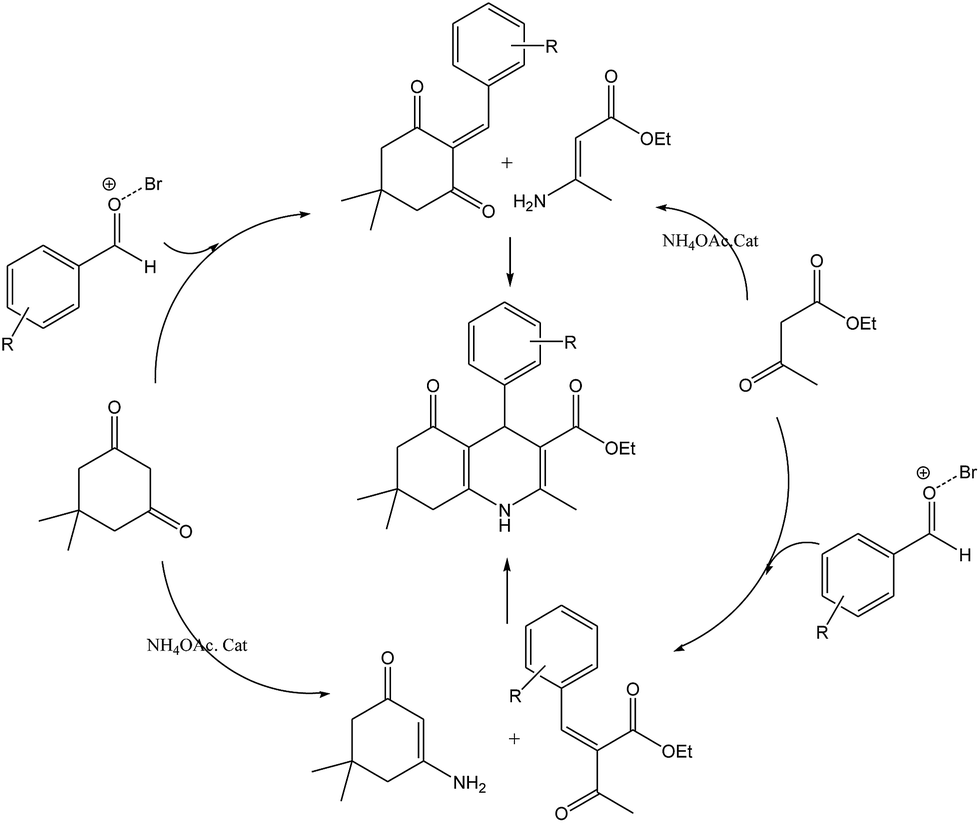 | ||
| Scheme 5 The proposed mechanism of synthesis of polyhydroquinoline derivatives by MNPs-TEDETA tribromide. | ||
2.4 Recyclability of the catalyst
The recyclability of the catalyst was studied using a model reaction between 2-aminobenzamide and 4-chlorobenzaldehyde in the presence of catalyst in the reflux of EtOH under the optimization conditions. After completion of the reaction, the product was dissolved in hot EtOH. Then the catalyst was separated from the product using an external magnet. Then EtOH was removed using evaporation. Finally, the pure product was obtained by recrystallizing if from the EtOH. The recycled catalyst was dried and used for further runs and its activity did not show any significant decrease even after six runs (Fig. 7).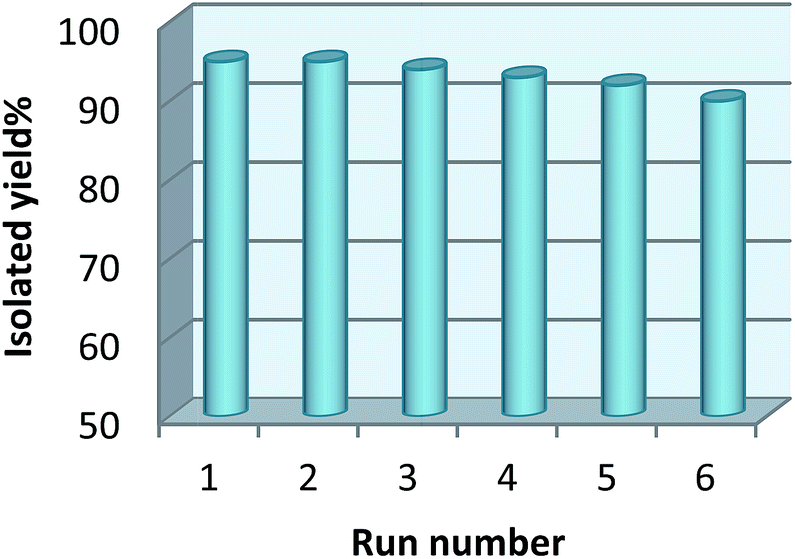 | ||
| Fig. 7 Recovery of the catalyst in the synthesis of 2,3-dihydroquinazolin-4(1H)-ones. | ||
2.5 Hot filtration
A hot filtration technique for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones was also studied. A hot filtration technique for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones was investigated, using 4-chlorobenzaldehyde (1 mmol), 2-aminobenzamide (1.05 mmol) and MNPs-TEDETA tribromide (0.05 g) and refluxing the mixture in EtOH. This reaction was carried out for 80 min and the product yield was 95%. The reaction was repeated and product yield in a reaction time of 40 min was 53%. In another reaction, the reaction repeated and after 40 min, the catalyst was separated by an external magnet and washed with hot EtOH, then the solution was allowed to go on reacting for the remaining 40 min with stirring. After purification, the product yield was 55%.2.6 Comparison of the catalysts
To demonstrate the advantages of MNPs-TEDETA tribromide, in comparison with other reported catalysts, the results of the preparation of 2-(3,4-dimethoxyphenyl)-2,3-dihydoquinazolin-4(1H)-one from 3,4-dimethoxybenzaldehyde and anthranilamide were compared with the previous reports in the literature. As shown in Table 5, these results indicated the efficiency of the proposed methodology in terms of the reaction yield and rate as compared to the results in the literature reports. The prepared catalyst in this work is a metal free catalyst.| Entry | Condition | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|
| a This work. | ||||
| 1 | CuCl2/Fe3O4-TEDETA (0.005 g), EtOH, reflux | 30 | 96 | 4 |
| 2 | GSA@MNPs (0.01 g), EtOH, reflux | 25 | 98 | 14 |
| 3 | BiBr3 (5 mol%), acetonitrile, RT | 30 | 84 | 18 |
| 4 | MES (10 mol%), EtOH, 60 °C/microwave irradiation | 180/12 | 86/88 | 19 |
| 5 | SiO2–FeCl3 (0.005 g), solvent free, 80 °C | 42 h | 90 | 20 |
| 6 | Boehmite-SSA (0.03 g), EtOH, reflux | 130 | 88 | 21 |
| 7 | Boehmite-Si-DSA (0.03 g), EtOH, reflux | 60 | 96 | 22 |
| 8 | MNPs-TEDETA tribromide (0.05 g), EtOH, 80 °C | 35 | 99 | a |
3 Conclusions
In summary, MNPs-TEDETA tribromide were synthesized for use as a new, reusable and efficient catalyst for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones by the condensation between various aldehydes and anthranilamide in the presence of MNPs-TEDETA tribromide and EtOH at 80 °C. The MNPs-TEDETA tribromide were also investigated for use in the synthesis of polyhydroquinoline via a one-pot, multicomponent condensation reaction between aldehyde, dimedone, EtOAc, ammonium acetate and MNPs-TEDETA tribromide as catalyst in PEG at 80 °C. The benefits of the catalyst are its novelty, easy preparation, functionalization without the necessity of difficult conditions, easy separation by an external magnet, and low toxicity and price. Also the catalyst can be reused six times without any significant loss of its activity.4 Experimental
4.1 Preparation of the magnetic Fe3O4 nanoparticles (MNPs)
A mixture of FeCl3·6H2O (5.858 g) and FeCl2·4H2O (2.221 g) was dissolved in 100 mL of deionized water until the salts dissolved completely. Then, 10 mL of 30% NH4OH was added in to the reaction mixture under a N2 atmosphere for about 30 min at 80 °C with vigorous mechanical stirring. NPs of Fe3O4 were collected and washed with doubly distilled water (five times).134.2 Preparation of MNPs coated with 3-chloropropyltrimethoxysilane (MNPs-CPTMS)
CPTMS (1.5 mL) was added to the magnetic Fe3O4 nanoparticles (0.5 g) in 50 mL EtOH/H2O (1:1) under a N2 atmosphere at 40 °C for about eight hours. Then, the final product was separated using an external magnet. For removal of the unattached substrates, the product was washed with EtOH and then dried at room temperature (RT).
4.3 Preparation of MNPs-TEDETA tribromide
TEDETA (2 mmol) was added to Et3N (4 mmol) at RT for 30 minutes. The MNPs-CPTMS were added to the mixture in dry toluene under reflux conditions for 24 h. MNPs-TEDETA were prepared after washing with deionized water and EtOH and dried overnight. In the next step, HBr (47%) was added to the MNPs-TEDETA and the mixture was stirred for an hour. Ultimately Br2 in carbon tetrachloride was added to the mixture and stirred for 6–7 h. Then the MNPs-TEDETA tribromide was collected using an external magnet and washed with doubly distilled water and EtOH.4.4 General procedure for the synthesis of 2,3 dihydroquinazolin-4(1H)-ones derivatives
A stirred mixture of aldehyde (1 mmol), 2-aminobenzamide (1.05 mmol) and MNPs-TEDETA tribromide (0.05 g), was reacted under reflux with EtOH. After the completion of the reaction, which was monitored using thin-layer chromatography (TLC), the product was dissolved in hot EtOH, and then the catalyst was separated from the product using an external magnet. Then EtOH was removed by evaporation. Finally, the pure products were obtained by recrystallization in EtOH.4.5 General procedure for the synthesis of polyhydroquinoline derivatives
A mixture of aldehyde (1 mmol), dimedone (1 mmol), EtOAc (1 mmol), ammonium acetate (1.2 mmol) and MNPs-TEDETA tribromide (0.05 g) was stirred in PEG at 80 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, the catalyst was separated using an external magnet and then product was extracted with ethyl acetate. Finally, the solvent was evaporated and all the products were recrystallized in EtOH, and the pure products were obtained in good to excellent yields.4.6 Characterization data of all compounds
Acknowledgements
The financial support for this research work from the Ilam University, Ilam, Iran is gratefully acknowledged.References
- J. Govan and Y. K. Gun’ko, Nanomaterials, 2014, 4, 222 CrossRef CAS.
- A. Naghipour and A. Fakhri, Catal. Commun., 2016, 73, 39 CrossRef CAS.
- A. Ghorbani-Choghamarani and M. Norouzi, Appl. Organomet. Chem., 2016, 30, 140 CrossRef CAS.
- A. Ghorbani-Choghamarani and M. Norouzi, J. Mol. Catal. A: Chem., 2014, 395, 172 CrossRef CAS.
- Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N. S. Hosmane, ChemCatChem, 2010, 2, 365 CrossRef CAS.
- A. Rostami, B. Tahmasbi, F. Abedi and Z. Shokri, J. Mol. Catal. A: Chem., 2013, 378, 200 CrossRef CAS.
- M. Ghorbanloo, R. Tarasi, J. Tao and H. Yahiro, Turk. J. Chem., 2014, 38, 488 CrossRef CAS.
- A. Rostami, B. Tahmasbi, H. Gholami and H. Taymorian, Chin. Chem. Lett., 2013, 24, 211 CrossRef CAS.
- M. Hajjami, A. Ghorbani-Choghamarani and F. Gholamian, Bulg. Chem. Commun., 2015, 47, 119 Search PubMed.
- B.-H. Chen, J.-T. Li and G.-F. Chen, Ultrason. Sonochem., 2015, 23, 59 CrossRef CAS PubMed.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Struct., 2014, 1072, 173 CrossRef CAS.
- M. Wang, T. T. Zhang, Y. Liang and J. J. Gao, Monatsh. Chem., 2012, 143, 835 CrossRef CAS.
- A. Ghorbani-Choghamarani and G. Azadi, RSC Adv., 2015, 5, 9752 RSC.
- M. Hajjami and B. Tahmasbi, RSC Adv., 2015, 5, 59194 RSC.
- B. Zhang, L. Shi and R. Guo, Catal. Lett., 2015, 145, 1718 CrossRef CAS.
- A. Saffar-Teluri and S. Bolouk, Monatsh. Chem., 2010, 141, 1113 CrossRef CAS.
- Z. Karimi-Jaberi and R. Arjmandi, Monatsh. Chem., 2011, 142, 631 CrossRef CAS.
- K. R. Gopinath, H. S. Shekar, K. J. Rajendraprasad, H. Nagabhushana and M. Krishnappa, World J. Pharm. Pharm. Sci., 2016, 5, 1272 CAS.
- V. B. Labade, P. V. Shinde and M. S. Shingare, Tetrahedron Lett., 2013, 54, 5778 CrossRef CAS.
- M. Ghashang, K. Azizi, H. Moulavi-Pordanjani and H. R. Shaterian, Chin. J. Chem., 2011, 29, 1617 CrossRef.
- A. Ghorbani-Choghamarani and B. Tahmasbi, New J. Chem., 2016, 40, 1205 RSC.
- M. Hajjami, A. Ghorbani-Choghamarani, R. Ghafouri-Nejad and B. Tahmasbi, New J. Chem., 2016, 40, 3066 RSC.
- A. Davoodnia, S. Allameh, A. R. Fakhari and N. Tavakoli-Hoseini, Chin. Chem. Lett., 2010, 21, 550 CrossRef CAS.
- A. Rostami, Y. Navasi, D. Moradi and A. Ghorbani-Choghamarani, Catal. Commun., 2014, 43, 16 CrossRef CAS.
- S. Otokesh, E. Kolvari, A. Amoozadeh and N. Koukabi, RSC Adv., 2015, 5, 53749 RSC.
- M. Hajjami and S. Kolivand, Appl. Organomet. Chem., 2016, 30, 282 CrossRef CAS.
- A. Ghorbani-Choghamarani, Z. Darvishnejad and B. Tahmasbi, Inorg. Chim. Acta, 2015, 435, 223 CrossRef CAS.
- B. Atashkar, A. Rostami and B. Tahmasbi, Catal. Sci. Technol., 2013, 3, 2140 CAS.
- A. Ghorbani-Choghamarani, B. Tahmasbi, P. Moradi and N. Havasi, Appl. Organomet. Chem., 2016, 30, 619 CrossRef CAS.
- H. Cao, J. He, L. Deng and X. Gao, Appl. Surf. Sci., 2009, 255, 7974 CrossRef CAS.
- M. Singh and N. Raghav, Bioorg. Chem., 2015, 59, 12 CrossRef CAS PubMed.
- F. Havasi, A. Ghorbani-Choghamarani and F. Nikpour, Microporous Mesoporous Mater., 2016, 224, 26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15474c |
| This journal is © The Royal Society of Chemistry 2016 |