
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFriend or foe? The role of solvents in non-triplet, intraligand charge transfer sensitization of lanthanide(III) luminescence†
Wai-Sum
Lo
,
Wing-Tak
Wong
* and
Ga-Lai
Law
*
Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: w.t.wong@polyu.edu.hk; ga-lai.law@polyu.edu.hk
First published on 27th July 2016
Abstract
Sensitization of lanthanide luminescence in the visible and near infra-red (NIR) region was achieved via a non-triplet energy transfer pathway by long-wavelength excitation of an intraligand charge transfer (ILCT) band of a diethylamino-based ligand system. A comprehensive photophysical and solvatochromic study is conducted to evaluate the luminescence properties of our Eu(III), Sm(III) and Yb(III) complexes. The effects of solvent coordination and solvent quenching on lanthanide luminescence were studied in detail and efforts were also devoted into examining the ambiguous energy transfer mechanism of Yb(III) sensitization.
Introduction
Trivalent lanthanides (Ln(III)) are a “stubborn” but fascinating group of luminescent centers. Stubborn as in their inflexibility to absorb light, with very low extinction coefficients (ε ≈ 1–10 L mol−1 cm−1),1 which arises due to the Laporte-forbidden f–f transitions. Therefore direct excitation is neither practical nor feasible in extensive applications as it often requires high-power lasers. Nevertheless, the forbidden nature imparts fascinating and unique luminescence characteristics to the Ln(III), such as long luminescence lifetimes (μs to ms), fingerprint emission profiles, fine emission bands and large pseudo-Stokes shifts. These properties have allowed lanthanides to reap benefits that organic chromophores and luminescent transition metal complexes lack and hence be applied in applications such as in displays and as optical imaging agents.1The antenna effect is a general term describing the use of a chromophore to harvest light energy and transfer it to the Ln(III), followed by subsequent radiative decay to yield f–f transitions and has become a common strategy for sensitizing lanthanide(III) luminescence. There are several energy transfer pathways from the chromophore to the Ln(III) center, with the most common one involves an intersystem crossing process – facilitated by heavy atom effect of Ln(III) – to populate the triplet excited state followed by energy transfer to the Ln(III) accepting state(s).1 The long-lived triplet state allows more time for the excited energy to be transferred to the Ln(III) but the energy difference between the photoexcitation and the accepting state(s) would have to be large to accommodate for intersystem crossing. An energy pathway circumventing the triplet excited state would allow lower energy excitation and thus be more advantageous in various applications.
The energy transfer mechanism of the sensitization process is worthy of fundamental research interest, too, as the physical prerequisites for different non-mutually exclusive mechanisms – Förster2 and Dexter3 – is closely correlated to the structural design of the chromophore, such as spectral overlap in FRET, and Ln(III)-chromophore distances. The ability to understand and manipulate such relationship(s) would help establish suitable blueprints for designated applications. The role of luminescence quenchers such as high-energy O–H, C–H oscillators with harmonics of resonant energies with the ΔE of Ln(III) should also be taken into account when designing the complex.4
In this work, we nominated three Ln(III) for our investigation: Eu(III) is chosen for its bright luminescence in the visible region and its hypersensitivity transition for studying the surrounding environment;5 Sm(III) for having an accepting state of similar energy to Eu(III) and its ability to emit in both the visible and NIR region;6 and Yb(III) for its low-energy NIR emission and intriguing energy transfer pathway.6a,7 The three Ln(III) was complexed with the same ligand system which consists of three 2-thenoyltrifluoroacetonate (TTA) moieties – a known, good antenna that sensitizes all three Ln(III) through the triplet-mediated energy transfer pathway8 – and a neutral tridentate scaffold with a diethylamino moiety imparting intraligand charge transfer (ILCT) character, which is able to sensitize Eu(III)9 and Sm(III)10 luminescence via a non-triplet mediated pathway. This current work extends the scope of our previous studies, expanding the work with different Ln(III) emitters as well as with various solvents of different polarity in an attempt to investigate the various processes mentioned above.
Results and discussion
The complexes were synthesized as previously reported (Scheme 1),10 where ligand 1 and the Ln(tta)3·xH2O (either commercially available or as-synthesized), were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in methanol at 50 °C for 16 hours. The crude was dissolved in minimum amount of diethyl ether and precipitated by n-hexanes for three cycles to obtain the product as yellowish solids. Different Ln(III) complexes exhibit identical solubilities, notably insoluble only in n-hexanes, petroleum ether and water.
:1 stoichiometry in methanol at 50 °C for 16 hours. The crude was dissolved in minimum amount of diethyl ether and precipitated by n-hexanes for three cycles to obtain the product as yellowish solids. Different Ln(III) complexes exhibit identical solubilities, notably insoluble only in n-hexanes, petroleum ether and water.
 | ||
| Scheme 1 Synthesis of Ln-1. (a) (i) Mg, I2, THF; (ii) cyanuric chloride, THF, reflux, 8 h (b) potassium pyrazolide, THF, reflux, 16 h. (c) Ln(tta)3, MeOH, 50 °C, 12 hours. | ||
UV-vis absorption and excitation spectra
The absorption spectra of the complexes in solution displayed three absorption bands in the ultraviolet region. The highest energy band at ca. 260 nm is assigned as the π–π* transition of the dipyrazolyltriazine scaffold and the band with a maximum at ca. 340 nm is assigned as the π–π* transition of the TTA moiety. The ILCT absorption band, which shifts with solvent polarity, could be observed with a maximum ranging from ca. 350 nm to 420 nm. The polar ground state of the molecule (donor–acceptor nature of the diethylaniline and dipyrazolyltriazine ends) imparts negative solvatochromism properties to the compound. Polar solvents stabilizes the polar ground state and hence the ΔE becomes larger, resulting in a hypsochromic shift.11 The excitation of the ILCT band does not necessarily sensitize lanthanide(III) luminescence, though, as the fluorescence may not be at a suitable energy level for energy transfer to take place. We have compared the UV-vis absorption spectra and the excitation spectra of the complexes to illustrate this finding.As seen in the excitation spectra (Fig. 1 and ESI Fig. S1–S12, S25–S37 and S50–S61†), all the complexes have an excitation band with a maximum at ca. 340 nm, which has a nearly identical band shape and maximum with the absorption band of the TTA moiety, indicating antenna effect via the TTA. In non-polar solvents (benzene, toluene, carbon tetrachloride), an additional excitation band which is superimposable with the ILCT absorption band could be found, confirming sensitization via excitation of the ILCT band as well, which is a non-triplet energy transfer pathway.9 The difference between the intensities of the absorption and excitation implies that the excitation of the ILCT ligand is more efficient in sensitizing lanthanide luminescence than the three moieties of TTA. Similar observations could be obtained from the spectra of chloroform, dichloromethane and 1,2-dichloroethane as well, albeit with a consistent red-shift of the excitation peak maxima compared to the absorption peak maxima.
 | ||
| Fig. 1 Normalized absorption (black) and excitation (red) spectra of Eu-1 in benzene (λem = 620 nm). | ||
The absorption spectra in polar solvents, on the other hand, mostly presented one broad band which we assign as a merged band of the TTA moiety and the blue-shifted ILCT band (vide supra). However, the excitation band only revealed a single band which is from the TTA as its maximum is at ca. 340 nm. The results indicate that the ILCT band could not effectively sensitize the Ln(III) in polar environments.
The excitation spectra of the three complexes are very similar, as expected of the ligand comprising of the same chemical components. For Sm(III) complexes, the excitation spectra of the visible (4G5/2 → 6H9/2) and NIR (4G5/2 → 4F5/2) transitions are also nearly identical in spectral-terms of both band shape and intensities. The results are as expected as both transitions decay from the same excited 4G5/2 level, as typically excitation spectra only take in account of where the excited energy is from and not where it ends.
In the case of acetonitrile (Fig. 2), the ILCT absorption band is rather distinct (λmaxca. 420 nm), despite it being a polar solvent (μ = 3.92). It is due to the highly polarizing nature of acetonitrile (ε = 37.5) which increases the rate of charge transfer from the diethylamino end to the dipyrazolyltriazine end such that stabilization by solvent dipole – which does not reach an electrostatic equilibrium with the molecule instantaneously – is rendered insignificant. This also explains the shoulders in the absorption spectra of the complexes in acetone (ε = 20.7) and DMSO (ε = 46.7).
 | ||
| Fig. 2 Normalized absorption and excitation spectra of Sm-1 in acetonitrile (λem = 645 nm (vis), 950 nm (NIR)). | ||
Emission spectra
The complexes were excited at three different wavelengths (330, 350, 390 nm). Excitation at 330 and 350 nm should give consistent results as the involvement of ILCT sensitization should be minimum, whereas the TTA moieties would be irrelevant with excitation of the ILCT band at 390 nm. The trend provides comprehensive data for comparison of different sensitization moieties. By studying the emission spectra of our Eu(III) complex, the hypersensitive 5D0 → 7F2 transition were found to be different in different solvents, hinting at different coordination environments around the emitting Eu(III) ion.12 In non-polar or less polar solvents, the splitting pattern of the hypersensitive transition peaks are quite similar, regardless of the energy transfer pathway undertaken prior to radiative decay (Fig. 3). As the solvent becomes more polar, the donor atom of the solvent molecules with increased nucleophilicity coordinate to the Ln(III) center and displace the neutral tridentate triazine-based scaffold. This could be reflected by the inconsistency in the splitting patterns in polar solvents. As the solvent molecules coordinate onto the Eu(III) center, the coordination environment varied to different extents depending on the number of molecules coordinated as well as the steric effect imposed on the overall structure. | ||
| Fig. 3 Room temperature emission spectra of Eu-1 in benzene at 0.1 absorbance (455 nm long pass filter). | ||
In polar solvents, while the splitting patterns are consistent between 330 and 350 nm excitation, they differ when excited at 390 nm. We believe that it is due to the remaining unperturbed complex molecules that retained the tridentate ligand in their structures to give considerably weaker luminescence via ILCT excitation.
Of the 5D0 → 7F1 and 5D0 → 7F2 transitions of Eu(III), the former transition is primarily magnetic dipole in nature and is largely independent of the ligand environment, acting as an internal standard for comparison with the latter hypersensitive transition. The asymmetry ratio, defined as the ratio between the integrated intensities of the two transitions provides an additional parameter to understand the effect of solvent coordination12 (the asymmetry ratio is intrinsically high due the unrestricted coordination manner of the three TTA molecules). A big difference can be observed between the non-polar and polar solvents, especially methanol and DMSO with high coordination strength. However, it must also be mentioned that similar asymmetry ratios have no implications of similar coordination environments, i.e. does not rule out solvent coordination in this case. The asymmetry ratio when excited at 390 nm also reinforces our above postulate by evidencing two emitting species of different extents of asymmetry.
Similarly, the emission intensities of Sm-1 in non-polar solvents via excitation at the ILCT band are comparable with TTA excitation (Fig. 4). The same trend in intensities with Eu-1 is observed as well due to the similar energy levels of their excited states. Yet the intrinsically weak quantum efficiency of Sm(III),6,10 solvent quenching and masking by strong ligand fluorescence are reasons behind the absence of any observable Sm(III) emission profile in certain polar solvents. While the hypersensitive transition 4G5/2 → 6H9/2 is not as sensitive as that of Eu(III), the asymmetry ratio between it and the 4G5/2 → 6H5/2 transition – predominantly magnetic dipole in nature – gives similar information as that of the Eu(III) counterpart and the results are summarized in Table 1; the same trend and explanation apply.
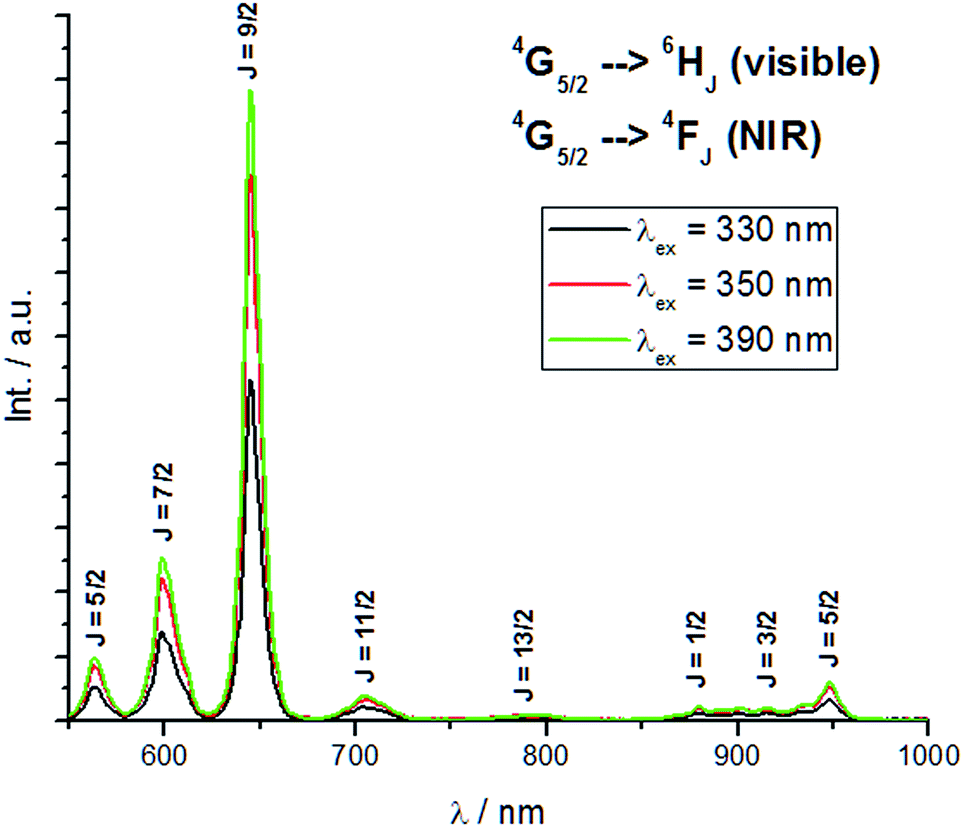 | ||
| Fig. 4 Room temperature emission spectra of Sm-1 in toluene at 0.3 absorbance (530 nm long pass filter). | ||
| Solvents | μ (D) | Eu-1 R a (λex = 350 nm) | Eu-1 R a (λex = 390 nm) | Sm-1 R b (λex = 350 nm) | Sm-1 R b (λex = 390 nm) | Eu-1 τ c | Sm-1 τ d | Eu-1 Φ e (%) | Sm-1 Φ e (%) |
|---|---|---|---|---|---|---|---|---|---|
| a I(5D0 → 7F2)/I(5D0 → 7F1). b I(4G5/2 → 6H9/2)/I(4G5/2 → 6H5/2). Blank entries are due to interference to transition intensities from ligand residual fluorescence (see ESI). c Emission lifetime of the 5D0 → 7F2 transition. d Emission lifetime of the 4G5/2 → 6H9/2 transition. e Relative to quinine sulfate in 0.1 M H2SO4, (λex = 350 nm; Φ = 0.577). f Instability upon dilution, inappropriate for measurement. Some of the SmIII data is taken from ref. 10. | |||||||||
| CCl4 | 0 | 18.4 | 18.5 | 10.8 | 12.8 | 0.494 | 0.159 | d | |
| Benzene | 0 | 19.5 | 19.5 | 10.6 | 10.2 | 0.432 | 0.106 | 23.8 | 3.75 |
| Benzene-d6 | 0 | — | 0.124 | — | 5.03 | ||||
| Toluene | 0.37 | 19.0 | 19.2 | 10.7 | 10.5 | 0.425 | 0.0935 | 30.6 | 3.13 |
| Toluene-d8 | 0.37 | — | 0.115 | — | 6.27 | ||||
| CHCl3 | 1.04 | 17.1 | 16.9 | 8.8 | 6.8 | 0.116; 0.510 | 0.027; 0.118 | f | |
| i-PrOH | 1.56 | 15.3 | — | 2.8 | — | 0.353 | 0.022 | 17.2 | 0.646 |
| CH2Cl2 | 1.6 | 14.3 | 12.4 | 5.4 | 3.8 | 0.013; 0.196 | 0.022; 0.121 | f | |
| CH3OH | 1.7 | 12.0 | — | 3.0 | — | 0.226 0.547 | 0.016 | 2.9 | 0.135 |
| CD3OD | 1.7 | 0.315; 1.109 | 0.131 | — | |||||
| THF | 1.75 | 16.6 | — | 5.3 | — | 0.383 | 0.027 | 10.7 | 1.21 |
| EA | 1.78 | 16.0 | 6.5 | 3.7 | — | 0.187; 0.397 | 0.059 | 7.1 | 1.36 |
| DCA | 1.8 | 17.1 | 11.6 | 4.6 | — | 0.114; 0.287 | 0.017; 0.092 | f | |
| Acetone | 2.88 | 16.6 | — | 2.7 | — | 0.290; 0.578 | 0.032; 0.110 | 7.1 | 0.572 |
| CH3CN | 3.92 | 19.6 | 6.7 | 4.7 | — | 0.618 | 0.056; 0.174 | 12.6 | 0.311 |
| DMSO | 3.96 | 10.0 | — | 2.1 | — | 0.752 | 0.026 | 12.9 | 0.481 |
The results are very similar for Yb-1, too. The unique emission profile from 900–1050 nm with a peak maximum at ca. 980 nm (Fig. 5), observed via both TTA and ILCT excitation, is attributed to the 2F5/2 → 2F7/2 transition, which is split into several components as a result of the ligand-field effect7 on both the emitting and ground states. Yb(III) luminescence via excitation of ILCT band in non-polar solvents were of comparable intensities to TTA excitation yet by comparing the absorption and excitation spectra (ESI, Fig. S50–S61†), the less smooth excitation curves – even in non-polar solvents – indicated very weak sensitization by antenna effect.
 | ||
| Fig. 5 Room temperature emission spectra of Yb-1 in toluene at 0.3 absorbance (830 nm long pass filter) showing the 2F5/2 → 2F7/2 transition. | ||
Energy transfer pathway
One of the main objectives of this work is to discover more about the ILCT band and the role it plays and could play in sensitizing Ln(III) luminescence. TTA is a well-known β-diketone capable of sensitizing Eu(III), Sm(III) and Yb(III) luminescence and the purpose of incorporating it in our complexes is to serve as an internal standard of the antenna effect. Low temperature (77 K) measurements of Gd-1 and Gd(tta)3 were done in 2-methyltetrahydrofuran in an attempt to measure the triplet states of the complex. Gd(III) was chosen as it is able to simulate the coordination environment of the other complexes – due to the similar ionic radii across the lanthanides – while eliminating the possibility of energy transfer to lanthanide(III) upon ligand excitation as its only excited state is located beyond 30000 cm−1. Excitation at 350 nm yielded a lower energy band (ESI, Fig. S74†) in Gd-1 with an emission lifetime of 3.6 ms. Comparison with Gd(tta)3 further confirms the assignment as the TTA triplet. An additional higher energy emission band with a maximum at ca. 400 nm was observed in which a biexponential lifetime was obtained (3.6 and 10 μs). When Gd-1 was excited at 390 nm (Fig. 6), the phosphorescence from TTA was absent and the same higher energy emission band (τ = 9.3 and 1.8 μs), assigned as the fluorescence of ILCT transition, was observed with a shoulder at ca. 422 nm (τ = 10 and 1.6 μs).
 | ||
| Fig. 6 Normalized room temperature absorption and emission spectra (λex = 390 nm) and low temperature excitation (λem = 400 nm) and emission (λex = 390 nm) spectra of Gd-1 in 2-MeTHF. | ||
As the antenna effect via the TTA moiety (triplet-mediated pathway) is well documented in literature, we would like to focus on the sensitization by excitation the ILCT band. A simplified Jablonski diagram is presented in Fig. 7 with the triplet energy level of TTA, the ILCT band and the accepting states of Eu(III), Sm(III) and Yb(III). The non-triplet ILCT excited state energy was determined to be ca. 23900 cm−1 by room temperature emission spectrum of Gd-1. The 5D2 and 5D1 accepting states of Eu(III) are at ca. 21500 and 19200 cm−1 respectively, and are seemingly more preferable destinations for energy transfer from the ILCT level due to a smaller energy gap,13 compared to 5D0 (ca. 17200 cm−1), although energy transfer to the latter should not be ruled out as the 4G5/2 accepting state (ca. 17900 cm−1) of Sm(III) is also a recipient of the excited energy in Sm-1. The energy of the excited state, however, is expected to change in different polarities. The less polar excited state will be stabilized to a larger extent than the polar ground state, thus reducing the gap with the accepting states, facilitating energy transfer. Considering other factors such as displacement of the ILCT ligand as well as quenching by oscillators of solvent molecules, various non-radiative processes dominate the energy transfer process hence resulting in the observed trend.
 | ||
| Fig. 7 Simplified Jablonski diagram depicting energy levels of chromophores and of LnIII. | ||
On the other hand, the spectral overlap between the ILCT band and the absorption of Yb(III) is minimal, especially without a lower energy triplet state. The energy transfer mechanism in the sensitization of Yb-1 may therefore be majorly a long range electron transfer process analogous to Horrocks Jr's work.7a Excitation of Yb-1 is believed to have caused an intramolecular electron transfer from the diethylamino moiety to the dipyrazolyltriazine and form a temporary charge-separated state: positive and negative charge at the donor and acceptor respectively. The extended Rehm–Weller equation14 could indicate whether this electron-transfer process is thermodynamically favorable:
| ΔGET = (Eox − Ered) − Es − eo2/εa |
Luminescence lifetimes and quantum yields
Quantum yields in chlorinated solvents were not measured as instability issues were observed.10,15 In chloroform, dichloromethane, dichloroethane, chlorobenzene, 1,2-dichlorobenzene and carbon tetrachloride, the complexes all suffer from an abnormal deterioration of their absorption spectra – either after prolonged idle time or dilution (Fig. S76–S79†). These solvents possess relative low dipole moments, and complexes at initial dissolution displayed the ILCT absorption bands. However, as time passes or, more severely, upon dilution (e.g. in relative quantum yield measurements), the ILCT band deteriorates – the ILCT absorbance plummets and λmax blue-shifts simultaneously – much quicker than the TTA part. Water was dismissed as a possible cause as the band deterioration problem was not observed in water-miscible solvents. The presence of hydrogen chloride, the main decomposition product in chlorinated solvents, was also suspected as the interference. The solvents were therefore pre-treated with different bases: sodium bicarbonate, potassium carbonate and calcium hydride. However, the problem persisted after the solvents were treated for 2 hours or overnight with the bases, therefore we decided to take the results in these solvents as reference only.Dipole moment was used as the parameter to relate solvents with their polarity and nucleophilicity. Under excitation of the TTA moiety, both Eu-1 and Sm-1 exhibit a general trend in which the quantum yields decreases with an increase of the dipole moment of the solvent molecules (Chart 1). The aromaticity of benzene and toluene provides rigidity to the ligand skeleton and minimizes vibronic deactivation, protecting the Ln(III) center well.
 | ||
| Chart 1 Visible luminescence quantum yield of Eu-1 and Sm-1 (vis) relative to quinine sulfate in 0.1 M H2SO4 (λex = 350 nm, Φ = 0.577) and NIR luminescence quantum yields of Sm-1 and Yb-1 in various solvents relative to Yb(tta)3(H2O)2 in toluene (λex = 340 nm, Φ = 0.35%) (estimated error is ±15%). Values of visible luminescence quantum yield of Sm-1 are taken from ref. 10. | ||
The solvent molecules interacting with the Ln(III) in the first coordination sphere, though, play an equally significant role. They could readily quench the luminescence via high energy vibrational overtones of O–H and C–H oscillators. The quenching effect is commonly explained by the energy gap law (EGL),16 which denotes the nonradiative quenching by vibrational overtones of high energy oscillators matching well with the energy gap between the excited state and the next lower energy state of the Ln(III). This multiphonon energy transfer process is analogous to the Förster resonance energy transfer and thus exhibits a distance-dependent relationship (α 1/r6). The EGL is prevalently applied to explain the quenching lanthanide luminescence since the electronic states of the lanthanides remain unaffected in different environments.1
To investigate such effect, lifetime measurements were done in selected deuterated solvents (Table 2). Some of the data in non-deuterated solvents for visible luminescence lifetimes of Sm-1 could be found in our previous publication.10 (It should also be noted that only the shorter lifetime the bi-exponential lifetimes were reported in ref. 10 as we did not look into the instability issues at that time.) Since the NIR luminescence of Sm-1 and Yb-1 are too weak for lifetime measurements, their quantum yields were measured instead (Chart 1), as quantum yield and luminescent lifetime are in a proportional relationship (Φ = τkr).
| Solvent | μ (D) | Sm-1 Φ a (%) | Yb-1 Φ b (%) |
|---|---|---|---|
| a Relative to Yb(tta)3 in toluene (λex = 340 nm, Φ = 0.35%). b Luminescence intensity too weak to give a satisfactory curve for calculations upon dilution. c Instability upon dilution, inappropriate for measurement. | |||
| Benzene | 0 | 6.09 | 0.43 |
| Benzene-d6 | 0 | 2.69 | 0.52 |
| Toluene | 0.37 | 4.72 | 1.22 |
| Toluene-d8 | 0.37 | 3.42 | 0.99 |
| i-PrOH | 1.56 | 0.71 | 0.05 |
| Fluorobenzene | 1.66 | 5.33 | 0.18 |
| CH3OH | 1.7 | b | 0.02 |
| CH3OD | 1.7 | 0.12 | 0.34 |
| CD3OD | 1.7 | 1.37 | 1.05 |
| THF | 1.75 | 0.97 | 0.20 |
| EA | 1.78 | 1.43 | 0.57 |
| Acetone | 2.88 | 0.77 | 0.07 |
| CH3CN | 3.92 | 0.69 | 0.31 |
| CD3CN | 3.92 | c | 1.74 |
| DMSO | 3.96 | b | 0.52 |
Methanol and acetonitrile molecules are expected to coordinate to the Ln(III) center and therefore in methanol-d4 and acetonitrile-d3 (for Yb-1 only, as instability issues similar to the chlorinated solvents were observed in repeated trials of Sm-1), the quantum yields are greatly enhanced from non-deuterated solvents. In methanol-d1 (CH3OD), the quantum yields of both complexes increased as well but were still some way off from methanol-d4. These results proved that, for donor molecules in the first coordination sphere, the presence of O–H oscillators quenches the luminescence but the extent of quenching by proximal C–H oscillators is more pronounced. The energy gaps between the emitting state and the next closest ground state of the Ln(III) are: ΔE(4G5/2 → 6F11/2) of Sm(III): ≈7400 cm−1 and ΔE(2F5/2 → 2F7/2) of Yb(III): ≈10200 cm−1. The drastic increase (>15×) in quantum yields of Yb-1 by deuteration of O–H in methanol can be explained by eliminating the efficient multiphonon quenching by the second O–H vibrational overtone (≈10500 cm−1), justifying the EGL. The relatively slight increase (3× in CD3OD and 6× in CD3CN) by deuteration of C–H is due to the less efficient matching of the third C–H overtone (≈11600 cm−1) with ΔE.
The quantum yield results of Sm-1, however, could not fit in the EGL preposition. The energy gap of Sm(III) did not match any overtones of the oscillators. The second and third aliphatic C–H overtones are at ≈8700 cm−1 and ≈11600 cm−1 respectively and only the first O–H overtone at ≈7000 cm−1 is close to matching with the energy gap. Deuteration of O–H did not give a significant increase as the second overtone of O–D vibration (≈7800 cm−1) matches quite well with the energy gap, too. Seitz and coworkers17 have already suggested that the EGL is not ‘universally relevant’ as the sole consideration of the smallest energy gap in the quenching process neglects the Franck–Condon principle, which governs the intensity of vibronic transitions – the exact nature of the quenching process discussed herein. This is particularly important in Ln(III) systems with multiple electronic states such as Sm(III), compared to Yb(III) which only has two states.
As our results suggest, the smallest energy gap is not effectively quenched by multiphonon relaxation in Sm-1 but the energy difference between the excited 4G5/2 and 6F9/2 states indeed matches well with the second C–H (≈8700 cm−1) and third C–D overtones (≈8400 cm−1). Comparing the quantum yields of CD3OD and CH3OD, though, still revealed a 10-fold difference and we believe it is due to the higher probability of having three quanta of C–H vibrational energy in resonance with ΔE(4G5/2 → 6F9/2) than four C–D vibrational quanta.18
The EGL was simply not comprehensive enough to estimate the quenching processes in Sm(III) compounds due to the many possibilities of vibronic transitions amongst the various electronic states. Furthermore, the NIR transitions are of lower energies than visible light transitions therefore the overtones required to match the energy differences are less, resulting in more probable quenching, hence the generally lower quantum yields in polar solvents.
The quantum yields obtained in fluorobenzene are also quite good for both complexes. The aromaticity increases the rigidity for the complex and the absorption band deterioration problem was not observed, too, as the C–F bond is less prone to decomposition than the C–Cl bond. The introduction of a highly electronegative fluorine atom also rendered the solvent molecule quite polar (comparable dipole moment with methanol), yet solvent coordination to the Ln(III) center is not expected due to steric hindrance of the aromatic ring and, therefore, the quantum yields are quite good as the Ln(III) centers are well-shielded from oscillators.
An interesting finding was presented in deuterated benzene and toluene measurements of Sm-1 and Yb-1 (Table 2). The NIR luminescence quantum yields exhibited a decrease, contrary to the general notion that deuteration of oscillators often results in less quenching. However, for visible luminescence of Sm-1, the quantum yield increased as expected, intriguing us to surmise that the deuteration of aromatic C–H leads to a preferential radiative decay of the excited energy at the 4G5/2 energy state of Sm-1 to the higher ΔE transitions, resulting in higher quantum yields than the NIR ones, although this remain a wild hypothesis.
The sensitization efficiency (ηsens) is a parameter that could be introduced to summarize the various processes that took place between photo-absorption and luminescence. It is calculated as the ratio between the intrinsic quantum yield (QLnLn, luminescence quantum yield by f–f-direct excitation) and the overall quantum yield (QLnL, via antenna effect). While the overall quantum yield can be obtained by conventional methods such as the relative and absolutely methods, measuring the intrinsic quantum yield is impractical, though technically feasible. Fortunately, the intrinsic quantum yield could be obtained from observed and radiative lifetimes. The observed lifetime is the lifetime measured experimentally and the radiative lifetime denotes the lifetime in the absence of any non-radiative deactivation pathways. Below are the equations for calculating the sensitization efficiency and, for europium(III), the radiative lifetime, obtained by theoretical methods that are detailed in ref. 19:
| ηsens = QLnL/QLnLn = (QLnL)(τrad)/τobs |
| 1/τrad = AMD,0n3(Itot/IMD) |
The sensitization efficiencies in various solvents of Eu-1 is presented in Table 3 and assuming Sm-1 and Yb-1 are isostructural, the results can be used as reference. Surprisingly, the intrinsic quantum yields of acetonitrile and DMSO were higher than that of the non-polar solvents. The poor sensitization efficiency, however, is similar to methanol and this corroborates with the quenching by coordinated solvent molecules mentioned previously.
| Solvent | μ (D) | Q LnLn (%, TTA) | η sens (%, TTA) | Q LnLn (%, ILCT) | Q LnL (%, ILCT) | η sens (%, ILCT) |
|---|---|---|---|---|---|---|
| a TTA: values calculated with overall quantum yield obtained by relative method against quinine sulfate (vide supra). ILCT: Values calculated with overall quantum yield obtained by absolute method using an integrating sphere with excitation at the absorption maxima of the ILCT band >400 nm to avoid second order of excitation light overlapping with 5D0 → 7FJ transitions. | ||||||
| CCl4 | 0 | — | — | 43 | 31 | 75 |
| Benzene | 0 | 40 | 59.5 | 53 | 32 | 61 |
| Toluene | 0.37 | 37 | 82.6 | 51 | 30 | 59 |
| CHCl3 | 1.04 | — | — | 46 | 18 | 37 |
| i-PrOH | 1.56 | 29 | 59.2 | — | — | — |
| CH2Cl2 | 1.6 | — | — | 27 | 6 | 22 |
| CH3OH | 1.7 | 13.7 | 21.2 | — | — | — |
| THF | 1.75 | 33 | 32.4 | — | — | — |
| EA | 1.78 | 13.6 | 52 | — | — | — |
| Acetone | 2.88 | 23 | 31 | — | — | — |
| CH3CN | 3.92 | 53.8 | 23.4 | — | — | — |
| DMSO | 3.96 | 52.9 | 24.4 | — | — | — |
Moderately polar solvents have understandably decent intrinsic quantum yields and sensitization efficiencies. For benzene and toluene, the intrinsic quantum yields are decent only, but the lack of solvent-associated deactivation pathways results in higher sensitization efficiencies which compensates for the overall quantum yields. Isopropanol, on the other hand, has a moderate sensitization efficiency despite the presence of O–H oscillators. We propose that it is a result of the relatively high viscosity of isopropanol that plays a role in hindering multiphonon quenching.
The efficiency of sensitization via the non-triplet energy transfer pathway was also investigated using an integrating sphere to measure the absolute quantum yields of Eu-1 in non-polar solvents as there is not a common quantum yield standard excited at ca. 400 nm, which is near the absorption maxima of the ILCT bands. Comparing between the intrinsic quantum yields of TTA and ILCT excitation in benzene and toluene, the values are 32% and 38% higher in ILCT excitation, confirming that the non-triplet sensitization pathway is more efficient in populating the excited states of Eu(III). Nevertheless, the shorter lifetime of the excited ILCT state compared to the excited triplet state of TTA would may be prone to competition from other singlet state deactivation pathways such as fluorescence.
Conclusion
In this paper, we investigated fundamental processes involved in the sensitization of visible and NIR luminescence of our Eu(III), Sm(III) and Yb(III) complexes with a ligand system containing 2-thenoyltrifluoroacetonates and a diethylaniline-based tridentate chromophore capable of imparting intraligand charge transfer character. This ligand system is able to sensitize all three Ln(III) and the photoluminescence studies of individual ions contributed to a library of findings with just one system. A detailed solvatochromic study was performed and the results were used to illustrate various processes during the antenna effect. The coordination of polar solvent molecules was evidenced in the hypersensitive transitions of Eu-1. Quantum yield measurements of Sm-1 offered further insights on the estimating luminescence quenching regarding the energy gap law and the sensitization efficiencies of Eu-1 were determined as well. Photoluminescence and cyclic voltammetry experiments of Yb-1 provided food for thought on the energy transfer mechanism of sensitizing Yb(III) luminescence via a non-triplet pathway by the ILCT band.Experimental
Materials and methods
2-Thenoyltrifluoroacetonate (tta) lanthanide salt Eu(tta)3·2H2O was purchased from Alfa Aesar. Sm(tta)3·2H2O and Yb(tta)3·2H2O were synthesized according to literature procedures with anhydrous SmCl3 (99.99%), anhydrous YbCl3 (99.99%) and 2-thenoyltrifluoroacetone (99%) from Aldrich. THF was dried with sodium metal and benzophenone. Spectrophotometric grade solvents, sulfuric acid concentrate and quinine sulfate were purchased from Sigma-Aldrich and deuterated solvents were purchased from Cambridge Isotope Laboratories and Sigma-Aldrich.Synthetic procedures
The synthesis of Sm-1 was reported in ref. 10 and is adopted for the syntheses of Eu-1 and Yb-1.:DCM/5:1. The product was further recrystallized in PE if it contains a blue-fluorescent purity under a handheld UV lamp excited at 254 nm. Yield: 57%. δH (400 MHz; CDCl3): 8.32 (m, 2H, ArH), 6.82 (m, 2H, ArH), 3.49 (q, J = 7.2 Hz, 4H, CH2), 1.25 ppm (t, J = 7.2 Hz, 6H, CH3). δC: 173.74, 170.87, 152.74, 132.54, 118.60, 111.02, 44.80, 12.57 ppm. MS (ESI+): 296 [(M − H)+], 319 [(M + Na)+].
:EA/100:1 to elute a yellow product. Yield: 48%. δH (400 MHz; CDCl3): 8.79 (m, 2H, ArH), 8.53 (d, 2H, py), 7.92 (s, 2H, py), 6.71 (d, 2H, py), 6.54 (m, 2H, ArH), 3.48 (q, J = 7.2 Hz, 4H, CH2), 1.24 ppm (t, J = 7.2 Hz, 6H, CH3). δC: 174.26, 162.67, 152.09, 144.92, 131.99, 130.16, 120.36, 110.66, 109.3, 44.65, 12.59 ppm. MS (ESI+): m/z 383 [(M + Na)+], 743 [(2M + Na)+].
:1 ratio in a solution of methanol and stirred at 50 °C overnight. The solvent was subsequently evaporated and the residue was re-dissolved in minimum amount of diethyl ether and precipitated by n-hexanes. The bright yellow complex was obtained by repeated precipitation thrice. Average yield: 80%.
Eu-1 . HRMS (ESI)+: m/z 955.0798 {[Eu(tta)2]+}. Elemental analysis (%) calcd for C47H43EuF9N8O6S3: C, 45.71; H, 3.51; N, 9.07. Found: C, 45.15; H, 3.08; N, 8.80.
Sm-1 . MS (ESI)+: m/z 954 {[Sm(tta)2]+}. Elemental analysis (%) calcd for C47H43SmF9N8O6S3: C, 45.77; H, 3.51; N, 9.08. Found: C, 45.89; H, 3.37; N, 8.91.
Yb-1 . MS (ESI)+: m/z 976 {[Yb(tta)2]+}. Elemental analysis (%) calcd for C47H43YbF9N8O6S3: C, 44.94; H, 3.45; N, 8.92. Found: C, 43.52; H, 3.12; N, 8.80.
Photophysical measurements
Solution samples of ca. 0.1 and 0.3 absorbances at 350 nm were prepared for visible and NIR photoluminescence measurements respectively. Measurements were prepared in the unit of absorbance instead of concentration as the relative absorbances at 350 nm are slightly different for the complexes in different solvents due to the ILCT character. The concentrations at 0.1 and 0.3 absorbances are estimated to be ca. 1.4 to 1.8 μM and 4.3 to 5.4 μM respectively. The concentrations are low enough to avoid inner filter effect. To avoid time-dependent solvent coordination and instability issues (Fig. S76–S79†), each sample solution was freshly prepared from solid sample. Separate samples were used for (1) UV-vis, emission and excitation scans; (2) luminescence lifetime measurements and (3) quantum yield measurements.All room temperature solution measurements were done in quartz cuvettes of 1 cm path length. UV-vis spectra were recorded with an HP UV-8453 spectrophotometer. Room temperature photoluminescence measurements data obtained with (1) Edinburgh Instruments FLSP920 spectrophotometer equipped with a Xe900 continuous xenon lamp (450 W), xenon flashlamp (60 W) and a Hamamatsu R928P cooled at −20 °C (for visible emission) and (2) PTI QuantaMaster™ 50 equipped with a 75 W xenon arc lamp, double emission monochromator using 400 nm blazed 1200 lines per mm or 1200 nm blazed 600 lines per mm gratings, a Hamamatsu R928 PMT (for visible emission) and a Hamamatsu R5108 PMT (for visible and/or NIR emission), both thermoelectrically cooled. Low temperature (77 K) measurements were measured on FLSP920 using an EPR dewar from Edinburgh Instruments. Samples were dissolved in 2-methyltetrahydrofuran, inserted into an EPR quartz sample rod and cooled with liquid nitrogen. Emission spectra were recorded at 30 min intervals until the intensity and emission profiles remained constant (≈2 hours) and the spectra were taken as final. Visible emission spectra obtained were corrected for the spectral responses and NIR emission spectra were not corrected due to the weak intensities and high noise levels.
Luminescence lifetimes and quantum yields
Luminescence lifetimes of visible emissions were measured with FLSP290 and fitted with Origin. Luminescence quantum yields were measured relative to quinine sulfate in 0.1 M sulfuric acid (λex = 350 nm, Φ = 0.577)21 for visible emission and to [Yb(tta)3(H2O)2] in toluene (λex = 340 nm, Φ = 0.35%)22 for NIR emissions. These standards were chosen as the excitation and emission wavelengths match well with our samples. Absolute quantum yields were measured using an integrating sphere from Edinburgh Instruments and the quantum yield values were obtained using the F900 software. All photophysical measurements were averages of triplicates.Cyclic voltammetry
Electrochemical study of the Sm-1 and Yb-1 complexes was performed on a CHI 1030 A instrument and the electrochemical cell was of conventional design. A glassy carbon disk (3 mm diameter) was used as working electrode, a Pt wire as counter electrode and a non-aqueous Ag/AgNO3 as reference electrode. All the electrochemical experiments were performed using 0.1 M tetrabutylammonium hexafluorophosphate (TBHP) in tetrahydrofuran and the solution was purged with argon. Ferrocene (Fc) was used as the internal standard, and all potentials are referenced to the ferrocene/ferrocenium (Fc/Fc+) couple. All scans were done at 100 mV s−1.Acknowledgements
We thank Dr L. Y. S. Lee and Dr C.-S. Tsang of our department for performing the cyclic voltammetry experiments, and for their useful discussion. Financial support from the Hong Kong Research Grants Council (PolyU 153012/15P), Hong Kong Polytechnic University (G-UC08 and 1-ZVGG), and the University Central Facility for Chemical and Environmental Analysis (UCEA) are gratefully acknowledged. WS Lo acknowledges the receipt of a postgraduate studentship from the Hong Kong Polytechnic University.References
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (b) E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed; (c) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC; (d) J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC; (e) L. Sun, X. Ge, J. Liu, Y. Qiu, Z. Wei, B. Tian and L. Shi, Nanoscale, 2014, 6, 13242–13252 RSC.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence - Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Spring, Verlag, Berlin, Heidelberg, 2011, vol. 7, pp. 1–46 Search PubMed.
- (a) F. R. Gonçalves e Silva, O. L. Malta, C. Reinhard, H.-U. Güdel, C. Piguet, J. E. Moser and J.-C. G. Bünzli, J. Phys. Chem. A, 2002, 106, 1670–1677 CrossRef; (b) K. Lunstroot, P. Nockermann, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans and K. Driesen, Inorg. Chem., 2009, 48, 3018–3026 CrossRef CAS PubMed; (c) A. T. Bui, A. Grichine, S. Brasselet, A. Duperray, C. Andraud and O. Maury, Chem.–Eur. J., 2015, 21, 17757–17761 CrossRef CAS PubMed; (d) Y. Liu, L. Sun, J. Liu, Y.-X. Peng, X. Ge, L. Shi and W. Huang, Dalton Trans., 2015, 44, 237–246 RSC.
- (a) W. D. Horrocks Jr, J. P. Bolender, W. D. Smith and R. M. Supkowski, J. Am. Chem. Soc., 1997, 119, 5972–5973 CrossRef; (b) S. Faulkner, B. P. Burton-Pye, T. Khan, L. R. Martin, S. D. Wray and P. J. Skabara, Chem. Commun., 2002, 1668–1669 RSC; (c) R. F. Ziessel, G. Ulrich, L. Charbonnière, D. Imbert, R. Scopelliti and J.-C. G. Bünzli, Chem.–Eur. J., 2006, 12, 5060–5067 CrossRef CAS PubMed; (d) N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Inorg. Chem., 2009, 48, 2908–2918 CrossRef CAS PubMed; (e) T. Lazarides, M. A. H. Alamiry, H. Adams, S. J. A. Pope, S. Faulkner, J. A. Weinstein and M. D. Ward, Dalton Trans., 2007, 1484–1491 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- (a) C. Yang, L. Fu, Y. Wang, J. P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010–5013 CrossRef CAS PubMed; (b) L.-M. Fu, X.-C. Ai, M.-Y. Li, X.-F. Wen, R. Hao, Y.-S. Wu, Y. Wang and J.-P. Zhang, J. Phys. Chem. A, 2010, 114, 4494–4500 CrossRef CAS PubMed.
- W.-S. Lo, J. Zhang, W.-T. Wong and G.-L. Law, Inorg. Chem., 2015, 54, 3725–3727 CrossRef CAS PubMed.
- C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 4th edn, 2011 Search PubMed.
- D. Parker, Coord. Chem. Rev., 2000, 205, 109–130 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- P. Kadjane, L. Charbonnière, F. Camerel, P. P. Lainé and R. Ziessel, J. Fluoresc., 2008, 18, 119–129 CrossRef CAS PubMed.
- (a) R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS; (b) G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1963, 38, 1187–1203 CrossRef CAS.
- C. Doffek, J. Wahsner, E. Kreidt and M. Seitz, Inorg. Chem., 2014, 53, 3263–3265 CrossRef CAS PubMed.
- C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Güden-Silber, J. Lügger, C. Platas-Iglesias and M. Seitz, J. Am. Chem. Soc., 2012, 134, 16413–16423 CrossRef CAS PubMed.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346–1353 RSC.
- R. G. Charles and R. C. Ohlmann, J. Inorg. Nucl. Chem., 1965, 27, 255–259 CrossRef CAS.
- J. W. Eastman, Photochem. Photobiol., 1967, 6, 55–72 CrossRef CAS.
- S. B. Meshkova, Z. M. Topilova, D. V. Bolshoy, S. V. Beltyukova, M. P. Tsvirko and V. Y. Venchikov, Acta Phys. Pol., A, 1999, 95, 983–990 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15365h |
| This journal is © The Royal Society of Chemistry 2016 |