
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilising alternative modifications of α-olefin end groups to synthesise amphiphilic block copolymers†
Edward L.
Malins
a,
Carl
Waterson
b and
C. Remzi
Becer
*c
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
bInnospec Limited, Innospec Manufacturing Park, Oil Sites Road, Ellesmere Port, Cheshire, CH65 4EY, UK
cPolymer Chemistry Laboratory, School of Engineering and Material Science, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: r.becer@qmul.ac.uk
First published on 22nd July 2016
Abstract
Amphiphilic block copolymers comprised of polyisobutylene (PIB) and poly(2-(dimethylamino)ethyl methacrylate) (DMAEMA) have been synthesised. This has been achieved via two new synthetic processes. Firstly, a PIB macroinitiator was synthesised from PIB that is primarily terminated by an α-olefin, hydroboration/oxidation of the terminal α-olefin followed by esterification with α-bromoisobutyryl bromide furnished the desired macroinitiator. The PIB macroinitiator was then utilised to polymerise DMAEMA by SET-LRP at different block lengths of PDMAEMA. However, end fidelity of the PIB macroinitiator was low (60%) and as such synthesising a block of PDMAEMA with a predictable degree of polymerisation was not possible. PIB-b-PDMAEMA block copolymers were also synthesised by reactive coupling of functional homopolymers, primary amine terminated PDMAEMA and polyisobutenyl succinic anhydride (PIBSA). Variable molecular weight well-defined PDMAEMA was synthesised by SET-LRP using an azide bearing initiator followed by transformation of the azide end group to a primary amine. All primary amine functionalised PDMAEMA was then coupled with PIBSA to synthesise PIB-b-PDMAEMA, demonstrated by GPC. However, the capabilities of this synthetic approach was limited as more than one PDMAEMA may couple to the PIBSA, therefore not synthesising pure PIB-b-PDMAEMA copolymers.
Introduction
Polyisobutylene (PIB) is a versatile material that is often utilised as it exhibits many interesting and unique properties, such as biostability, biocompatibility, low-temperature flexibility, gas-barrier and energy damping, chemical, thermal, and oxidative stability.1 Therefore, materials that incorporate PIB such as PIB macromonomers,2,3 polymer networks, amphiphilic polymer co-networks (APCN)4–8 and block copolymers continue to attract much attention.9–12PIB based block copolymers, which are copolymers that contains at least one discreet block of pure IB, can typically be synthesised via four generic synthetic approaches; sequential monomer addition, site transformation of the PIB end group, macromolecular coupling of homopolymers and dual-site initiators. Sequential monomer addition can be achieved by direct addition of a second monomer following the living cationic polymerisation (LCP) of IB, a living PIB end group will initiate and subsequently polymerise said second monomer to furnish a block copolymer. Although this method is conceptually simpler it can be synthetically challenging as stringent reaction conditions are required and only monomers polymerisable via LCP are viable, such as styrene13 and vinyl ethers.14 Alternatively, the site transformation also utilises end groups present on PIB but is much more versatile as end group can be modified to a range of polymerisation initiators or mediators compatible with many polymerisations techniques; such as living anionic polymerisation,15 group transfer polymerisation,16 reversible addition fragmentation chain transfer (RAFT) polymerisation,17,18 atom transfer radical polymerisation (ATRP)19–21 and using the same initiators it may be possible to utilise single electron transfer living radical polymerisation (SET-LRP). This allows the polymerisation of other monomer classes like (meth)acrylates and (meth)acrylamides. For example, Ivan and co-workers have recently demonstrated the ability to transform a telechelic PIB into an ATRP initiator via hydroboration and esterification. This macroinitiator was then used to polymerise polyethylene glycol methacrylate (PEGMA) and then methyl methacrylate (MMA) to synthesise well defined ABCBA pentablock copolymers.22 Furthermore, Huang and co-workers have synthesised a PIB RAFT agent to mediate the polymerisation of 2-(diethylamino)ethyl methacrylate (DEAEMA) to synthesise well defined amphiphilic block copolymers that self-assemble and are pH responsive.23
Although this approach of site transformation drastically increases the versatility of PIB and potentially allows the synthesis for a limitless number of block copolymers. Synthesis of PIB macromolecular initiators/mediators can require many synthetic steps, specialised reagents and may subsequently be suitable for only one other polymerisation technique. Therefore, a more general methodology for synthesising PIB block copolymers can be achieved by utilising commercial sauces of functionalised PIB. Such a source of commercial PIB exists and is commonly known as “highly reactive” as the material is primarily end capped by a vinylidene.24 The vinylidene group is commonly reacted with maleic anhydride to yield poly(isobutenyl succinic anhydride)s (PIBSAs), which are imidised with primary amines to the corresponding poly(isobutenyl succinimide)s (PIBSIs).25,26 Furthermore, highly reactive PIB is used to alkylate phenol via a Friedel–Crafts alkylation to synthesise poly(isobutenyl phenol)s (PIBPs) that are precursors for the so called “Mannich type dispersants”.27,28
Herein, we report the synthesis of poly(isobutylene)-b-poly(2-(dimethylamino)ethyl methacrylate) (PIB-b-PDMAEMA) block copolymers via two separate synthetic approaches both starting from the same commercial source of PIB. Firstly, PIB was converted to a traditional ATRP style initiator via hydroboration and esterification of the vinylidene end group before SET-LRP of DMAEMA (Scheme 1). Secondly, primary amine functionalised PDMAEMA was synthesised via SET-LRP and Staudinger reaction of an α-terminal azide before being reactively coupled to PIBSA to form PIB-b-PDMAEMA.
 | ||
| Scheme 1 Synthesis of PIB-b-PDMAEMA block copolymers via two synthetic routes; PIB macroinitiator and reactive coupling of homopolymers. | ||
Experimental
Materials
Copper(II) bromide (Aldrich, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (Aldrich, 99%), borane tetrahydrofuran complex solution 1.0 M in THF (Aldrich), 2-bromoethanol (Aldrich, 95%), sodium azide (Aldrich, >99.5%) and α-bromoisobutyryl bromide (Aldrich, 98%) were used as received. “Highly reactive” PIB and PIBSA was donated by Innospec LTD and used as received. 2-(Dimethylamino)ethyl methacrylate (Aldrich, 98%) was purified by passing through a short column of basic aluminium oxide prior to polymerisation. Copper(0) wire (Cormax, 0.25 mm, 500 g) was activated by washing with concentrated sulfuric acid prior to polymerisation.Instruments
Synthesis of PIBBiB
Borane tetrahydrofuran complex solution 1 M in THF (3.0 mL, 3.00 mmol, 1.2 eq.) and degassed n-hexane (10 mL) were added to a degassed flask. “Highly reactive” PIB (2.685 g, 2.45 mmol, 1 eq.) was dissolved in n-hexane (10 mL), degassed by gentle bubbling of N2 gas for 30 minutes and added drop wise to the borane solution over 30 minutes at 0 °C. Mixture was maintained at 0 °C for a further 5 hours. Sodium hydroxide solution (0.5 M, 5 mL, 2.5 mmol, 1.02 eq.) was added drop-wise followed by 30% hydrogen peroxide solution (0.5 mL, 4.4 mmol, 1.8 eq.) and left for 2 further hours. Distilled water (40 mL) and n-hexane (20 mL) were added to the reaction mixture. Organic layer was separated and washed with distilled water (3 × 40 mL), dried over magnesium sulfate and filtered before removing all n-hexane in vacuo.291H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.97–1.03 ((–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)3, PIB α-terminus), 1.05–1.16 ((–C3)2, PIB repeat unit), 1.39–1.46 (s, –C2–, PIB repeat unit), 1.79 (s, –C3, residual vinylidene), 2.00 (s, –C2–, residual vinylidene), 4.65 (s, –C
3)3, PIB α-terminus), 1.05–1.16 ((–C3)2, PIB repeat unit), 1.39–1.46 (s, –C2–, PIB repeat unit), 1.79 (s, –C3, residual vinylidene), 2.00 (s, –C2–, residual vinylidene), 4.65 (s, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CfHg, residual vinylidene), 4.86 (q, –CCHfg, residual vinylidene). FT-IR (neat): (cm−1) 3404 (br), 2949 (s), 2878 (s), 1470 (m), 1365 (s), 1231 (m), 1072 (m, C–O), 914 (w). GPC: Mn 1200 g mol−1, Mw 1900 g mol−1, Đ 1.56.
CfHg, residual vinylidene), 4.86 (q, –CCHfg, residual vinylidene). FT-IR (neat): (cm−1) 3404 (br), 2949 (s), 2878 (s), 1470 (m), 1365 (s), 1231 (m), 1072 (m, C–O), 914 (w). GPC: Mn 1200 g mol−1, Mw 1900 g mol−1, Đ 1.56.
Polyisobutylene hydroxyl (2.897 g, 2.4 mmol, 1 eq.) was dissolved in THF (20 mL) and degassed by gentle bubbling of N2 gas for 30 minutes, cooled to 0 °C before the addition of triethylamine (0.73 mL, 5.2 mmol, 2.2 eq.) and degassed for a further 10 minutes. α-Bromoisobutyryl bromide (0.65 mL, 5.2 mmol, 2.2 eq.) diluted with THF (20 mL) was degassed for 30 minutes before adding drop-wise to the polyisobutylene hydroxyl solution at 0 °C. Reaction temperature was maintained at 0 °C for a further 4 hours before subsequently reaching room temperature and stirring for 2 days. Reaction mixture was diluted with n-hexane (60 mL) and extracted with distilled water (2 × 60 mL) and methanol (60 mL). Organic layer was dried over magnesium sulfate and filtered before removing all n-hexane in vacuo.20,211H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.97–1.03 ((–C3)3, PIB α-terminus), 1.05–1.16 ((–C3)2, PIB repeat unit), 1.39–1.46 (s, –C2–, PIB repeat unit), 1.79 (s, –C3, residual vinylidene), 1.97 (s, –C(C3)2Br), 2.00 (s, –C2–, residual vinylidene), 4.65 (s, –CCfHg, residual vinylidene), 4.86 (q, –CCHfg, residual vinylidene). FT-IR (neat): (cm−1) 2947 (s), 2884 (s), 1819 (w), 1738 (m, CO), 1466 (s), 1365 (s), 1273 (w, C–O), 1230 (m), 1165 (m, C–O), 1113 (m, C–O), 1044 (w), 916 (w). GPC: Mn 1300 g mol−1, Mw 1900 g mol−1, Đ 1.44.
SET-LRP of DMAEMA using PIBBiB as macroinitiator
Cu(0) wire (5 cm), CuBr2, methanol, PMDETA and DMAEMA (1 mL, 5.93 mmol) (see Table 1 for molar equivalents of all reagents) were charged in that order into a Schlenk tube and degassed by gentle bubbling of N2 gas. The Schlenk tube was submerged into a water bath at 25 °C before the addition of degassed PIBBiB dissolved in toluene via degassed syringe. After 4 hours the remaining reaction mixture was diluted with THF and passed through a column of basic aluminium oxide to remove the catalyst. PIB-b-PDMAEMA was precipitated twice into n-hexane and dried in vacuo. 1H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.73–1.23 (m, –C3, PDMAEMA repeat unit, (–C3)2, PIB repeat unit, (–C3)3, PIB α-terminus), 1.33–1.47 (m, –C2–, PIB repeat unit), 1.66–2.11 (m, –C2–, PDMAEMA repeat unit), 2.17–2.36 (s, –N(C3)2), 2.49–2.70 (m, –C2N–), 3.91–4.18 (m, –COOC2–).
| Run | [DMAEMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PIBBiB]b:[PMDETA]:[Cu(II)Br2] :[PIBBiB]b:[PMDETA]:[Cu(II)Br2] |
DMAEMA Conv.a (%) | Crude | Precipitated | ||
|---|---|---|---|---|---|---|
| M n,GPC (g mol−1) | Đ | M n,GPC (g mol−1) | Đ | |||
| a Calculated from 1H-NMR spectroscopy. b PIBBiB Mn: 1300 g mol−1, Đ: 1.44. | ||||||
| P1 | 5:1:0.18:0.05 |
80 | 1600 | 2.15 | 3500 | 1.77 |
| P2 | 15:1:0.18:0.05 |
83 | 2500 | 3.50 | 7900 | 1.80 |
| P3 | 25:1:0.18:0.05 |
79 | 2900 | 3.43 | 8900 | 1.77 |
Synthesis of AEBiB
2-Bromoethanol (5.05 g, 0.04 mol, 1 eq.) and sodium azide (3.91 g, 0.06 mol, 1.5 eq.) were dissolved in a mixture of acetone (50 mL) and water (20 mL) before heating under reflux conditions at 60 °C for 18 hours. Acetone was removed in vacuo and the resulting aqueous layer was extracted with diethyl ether (5 × 20 mL). All organic layers were combined, dried over magnesium sulfate and filtered before removing all diethyl ether in vacuo.461H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 3.40 (t, J = 4.7, 5.2 Hz, N3C2CH2OH, 2H), 3.78 (t, J = 4.7, 5.1 Hz, N3CH2C2OH, 2H). FT-IR (neat): (cm−1) 3370 (br, m, O–H), 2940 (w), 2878 (w), 2093 (s, –NNN), 1633 (w), 1442 (w), 1284 (m), 1062 (m), 878 (m), 631 (m).
2-Azidoethanol (2.758 g, 0.032 mol, 1 eq.) was dissolved in THF (20 mL) and cooled to 0 °C before addition of TEA (5.3 mL, 0.038 mol, 1.2 eq.), followed by drop wise addition of α-bromoisobutyryl bromide (4.7 mL, 0.038 mol, 1 eq.) dissolved in THF (30 mL) over 2 hours, temperature was maintained at 0 °C for a further 2 hours before subsequently reaching room temperature and left to stand for 24 hours. THF was removed in vacuo and the crude product was dissolved in diethyl ether (30 mL). Organic layer washed with sat. NaHCO3 sol. (3 × 30 mL), dried over magnesium sulfate and filtered before removing all diethyl ether in vacuo. Product dissolved in THF and passed through a column of basic aluminium oxide before solvent was removed in vacuo.471H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 1.96 (s, (–C3)2, 6H) 3.53 (t, J = 4.9, 5.3 Hz, N3C2CH2–, 2H), 4.34 (t, J = 4.9, 5.1 Hz, N3CH2C2COO–, 2H). FT-IR (neat): (cm−1) 2977 (w), 2099 (s, –NNN), 1734 (s, CO), 1467 (m), 1269 (s), 1155 (s), 1107 (s), 1034 (w), 945 (w), 841 (w), 643 (w).
SET-LRP of DMAEMA using AEBiB as initiator
Cu(0) wire (10 mL), CuBr2, methanol (10 mL), PMDETA and DMAEMA (10 mL, 59.3 mmol) (see Table 2 for molar equivalents of all reagents) were charged in that order into a Schlenk tube and degassed by gentle bubbling of N2 gas. The Schlenk tube was then submerged into a water bath at 25 °C before the addition of degassed AEBiB via a degassed syringe. Samples were taken at desired time points via degassed syringe, diluted with THF and immediately passed through a short column of basic aluminium oxide to remove the catalyst. After 4 hours the remaining reaction mixture was diluted with THF and passed through a column of basic aluminium oxide. PDMAEMA was precipitated twice into n-hexane and dried in vacuo. 1H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.78–1.17 (m, –C3), 1.66–2.11 (m, –C2–), 2.17–2.36 (s, –N(C3)2), 2.49–2.70 (m, –C2N–), 3.91–4.18 (m, –COOC2–). FT-IR (neat): (cm−1) 2946 (w), 2820 (w), 2768 (w), 2100 (w, –NNN, P2.09–P2.11 only), 1726 (s), 1455 (m), 1269 (m), 1147 (s), 1061 (m), 1016 (m), 965 (m), 853 (w), 747 (w), 543 (w).
| Run | [DMAEMA]:[AEBiB]:[PMDETA]:[Cu(II)Br2] |
DMAEMA Conv.a (%) | Before reduction | New run | After reduction | |||
|---|---|---|---|---|---|---|---|---|
| Mn,GPC (g mol−1) | Đ | Mn,GPC (g mol−1) | Mp,GPC (g mol−1) | Đ | ||||
| a Calculated from 1H-NMR spectroscopy. | ||||||||
| P4 | 5:1:0.18:0.05 |
78 | 2000 | 1.21 | P7 | 2100 | 2600 | 1.24 |
| P5 | 15:1:0.18:0.05 |
82 | 3400 | 1.20 | P8 | 3400 | 4500 | 1.24 |
| P6 | 25:1:0.18:0.05 |
85 | 4800 | 1.26 | P9 | 5400 | 6700 | 1.33 |
Reduction of N3-PDMAEMA to H2N-PDMAEMA
N3-PDMAEMA (1 eq.) and triphenylphosphine (1.2 eq.) were dissolved in THF (40 mL) and heated at 50 °C over night before the addition of distilled water (1 mL). Reaction heated at 50 °C for a further 2 hours. H2N-PDMAEMA was precipitated twice into n-hexane and dried in vacuo. 1H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.78–1.17 (m, –C3), 1.66–2.11 (m, –C2–), 2.17–2.36 (s, –N(C3)2), 2.49–2.70 (m, –C2N–), 3.91–4.18 (m, –COOC2–). FT-IR (neat): (cm−1) 2946 (w), 2820 (w), 2768 (w), 1726 (s), 1455 (m), 1269 (m), 1147 (s), 1061 (m), 1016 (m), 965 (m), 853 (w), 747 (w), 543 (w).
Macromolecular coupling of H2N-PDMAEMA
H2N-PDMAEMA (1 eq.) and PIBSA (1.25 eq.) were dissolved in toluene (50 mL) and heated at 90 °C for 2 hours before cooling to room temperature. PIB-b-PDMAEMA was precipitated twice into n-hexane and dried in vacuo. 1H-NMR (CDCl3, 300 MHz, 298 K): δ (ppm) 0.73–1.23 (m, –C3, PDMAEMA repeat unit, (–C3)2, PIB repeat unit, (–C3)3, PIB α-terminus), 1.33–1.47 (m, –C2–, PIB repeat unit), 1.66–2.11 (m, –C2–, PDMAEMA repeat unit), 2.17–2.36 (s, –N(C3)2), 2.49–2.70 (m, –C2N–), 3.91–4.18 (m, –COOC2–).
Results and discussion
PIB end-capped by a vinylidene group has been previously converted to an ATRP/SET-LRP macroinitiator. This has been achieved via hydroboration of the terminal vinylidene to a primary alcohol, ω-hydroxy polyisobutylene (PIBH), followed by esterification of said alcohol with BiBB.20,21,29 Macroinitiators of this nature have then been utilised at polymerising methacrylate monomers to yield the corresponding PIB block copolymers.20,21 Therefore, this methodology was utilised to functionalise commercial PIB to an ATRP/SET-LRP macroinitiator and subsequently polymerise DMAEMA via SET-LRP (Scheme 1).Hydroboration of PIB successfully converted 90% of the vinylidene to primary alcohol. 1H-NMR spectra of PIB and PIBH overlaid as well as a magnified vinylidene proton region show the disappearance of vinylidene protons (ESI Fig. S1†). Furthermore, commercial PIB contains a mixture of terminal olefin end groups, 1H-NMR allows the identification of two olefins contained in the PIB and calculation of the relative concentrations, 10:1 external:internal terminal olefin. Both terminal olefins were converted which results in a mixture of primary and secondary alcohols. Therefore, relative concentrations of PIBH primary:PIBH secondary are 9:1, calculated by relative decrease in olefin protons to PIB repeat unit. Following the hydroboration of PIB, PIBH was esterified to poly(isobutylene bromoisobutyrate) (PIBBiB). 1H-NMR confirms the structure by appearance of a signal at 1.97 ppm corresponding to the two methyl groups of the new initiating group (ESI Fig. S2†). Furthermore, GPC showed a slight increase in molecular weight as expected and a reduction in the polymer dispersity, Mn: 1300 g mol−1, Đ: 1.44.
SET-LRP has been shown to be an effective technique for polymerising more hydrophobic monomers by utilising a medium that is a mixture of polar and non-polar solvents,30–33 or a mixture of non-polar solvents and polar additives such as phenol.33,34 Therefore, SET-LRP of a polar monomer (DMAEMA) using a hydrophobic PIB macroinitiator is theoretically possible using a mixed solvent system of methanol, to achieve adequate polarity to disproportionate Cu(I)Br to Cu(0) and Cu(II)Br2, and toluene to solubilise the PIB (Scheme 1).
SET-LRP of DMAEMA using PIBBiB as initiator was successful at synthesising PIB-b-PDMAEMA. Monomer to initiator ratio was adjusted from 25:1 to 15:1 to 5:1 to synthesise three different molecular weight block copolymers varying only in the DP of the PDMAEMA block. DMAEMA conversion was ≥79% for each experiment and crude Mn measurements did increase as expected with increasing monomer to initiator ratio (Table 1). However, GPC traces of the crude block copolymers show very ill defined multi modal traces (Fig. 1). Furthermore, GPC traces show a significant low molecular weight shoulder that overlay well with PIBBiB. In reality this low molecular weight shoulder could correspond too many different species of PIB. A significant PIB impurity would be due to the commercial PIB not being 100% terminated by olefins and therefore containing PIB that is incapable of functionalisation. In conjunction with subsequent functionalisations; hydroboration and esterification, which did not proceed to full conversion would lead to the significant impurity of PIB seen in the final polymerisation.
 | ||
| Fig. 1 1H-NMR spectrum (CDCl3) of P1 (top left) crude and precipitated GPC traces of P1 (top right), P2 (bottom left), and P3 (bottom right) overlaid against PIBBiB. | ||
Fortunately, the block copolymers are insoluble in n-hexane, which makes it possible to remove the PIB impurity by precipitation. GPC of the precipitated PIB-b-PDMAEMA block copolymers show a dramatic increase in Mn as well as lowering dispersity (Table 1). Furthermore the final GPC chromatograms are now more symmetrical and modal with less low molecular weight polymer impurity (Fig. 1). An alternative synthetic methodology was required to synthesis PIB-b-PDMAEMA block copolymers that utilises a commercial source of PIB that could potentially minimise the limitations of utilising a PIB macroinitiator with reduced end group fidelity.
Cyclic anhydrides can be quickly and efficiently ring opened by a primary amine upon heating. Firstly, the amine ring opens the cyclic anhydride to form a new amide bond and a carboxylic acid. Given sufficiently high temperature (∼160–200 °C) and removal of water, the carboxylic acid and amide bond can cyclise with loss of water to form a cyclic imide. Reactions of cyclic anhydrides and secondary amines do not continue beyond the ring opening, forming an amide bond and a carboxylic acid. This chemistry has been exploited previously to functionalise monomer repeat units35–37 or polyolefin end groups.38–41 Another interesting application of this reaction is the possibility to couple two discreet homopolymers with complimentary end groups. Macosko et al. synthesised polystyrene (PS) with a range of functionalities; aliphatic primary amine, aromatic primary amine, cyclic anhydride, hydroxyl, carboxylic acid, epoxy and oxazoline.42 After heating reactive pairs of these functionalities at 180 °C for 2 min, they found aliphatic primary amine – cyclic anhydride to be superior, achieving >99% conversion compared to aromatic primary amine – cyclic anhydride (12.5%) and carboxylic acid – epoxy (9%), monitored by GPC.42 This coupling approach was utilised for the synthesis of PIB-b-PDMAEMA block copolymers by coupling an aliphatic primary amine functional PDMAEMA (H2N-PDMAEMA) to PIBSA.
H2N-PDMAEMA was obtained after reduction of an α-terminal azide present following polymerisation when using azidoethyl-2′-bromoisobutyrate (AEBiB) as initiator.43,44 DMAEMA was polymerised via SET-LRP utilising the same conditions utilised earlier, however the solvent system was neat methanol as toluene was no longer required to dissolve the initiator (Scheme 1). Three different molecular weights of N3-PDMAEMA (P4–6) were synthesised with good control over polymer dispersity and high monomer conversion was achieved (Table 2). Furthermore, the presence of the azide functionality can be confirmed via1H-NMR and FT-IR spectroscopy (Fig. 2).
 | ||
| Fig. 2 FT-IR spectrum (left) and 1H-NMR spectrum (CDCl3) (right) of P4 (black) and P7 (red). | ||
The Staudinger reaction has been commonly utilised as a simple and mild method of reducing an azide to a primary amines.45 Samples P4–6 were treated with triphenylphosphine and water to synthesise the corresponding NH2-PDMAEMA samples (P7–9). Reduction of the α-terminal azide to primary amine increased both the Mn and dispersity of samples (Table 2). Furthermore, GPC traces show a slight high molecular weight shoulder now present after the reduction (Fig. 3). This shoulder may have occurred due to a potential coupling reaction between the newly formed primary amine and any residual alkyl bromide found on the ω-terminus that was retained throughout polymerisation. Such coupling reactions have been demonstrated between small molecule primary amines and polymers prepared by ATRP.45
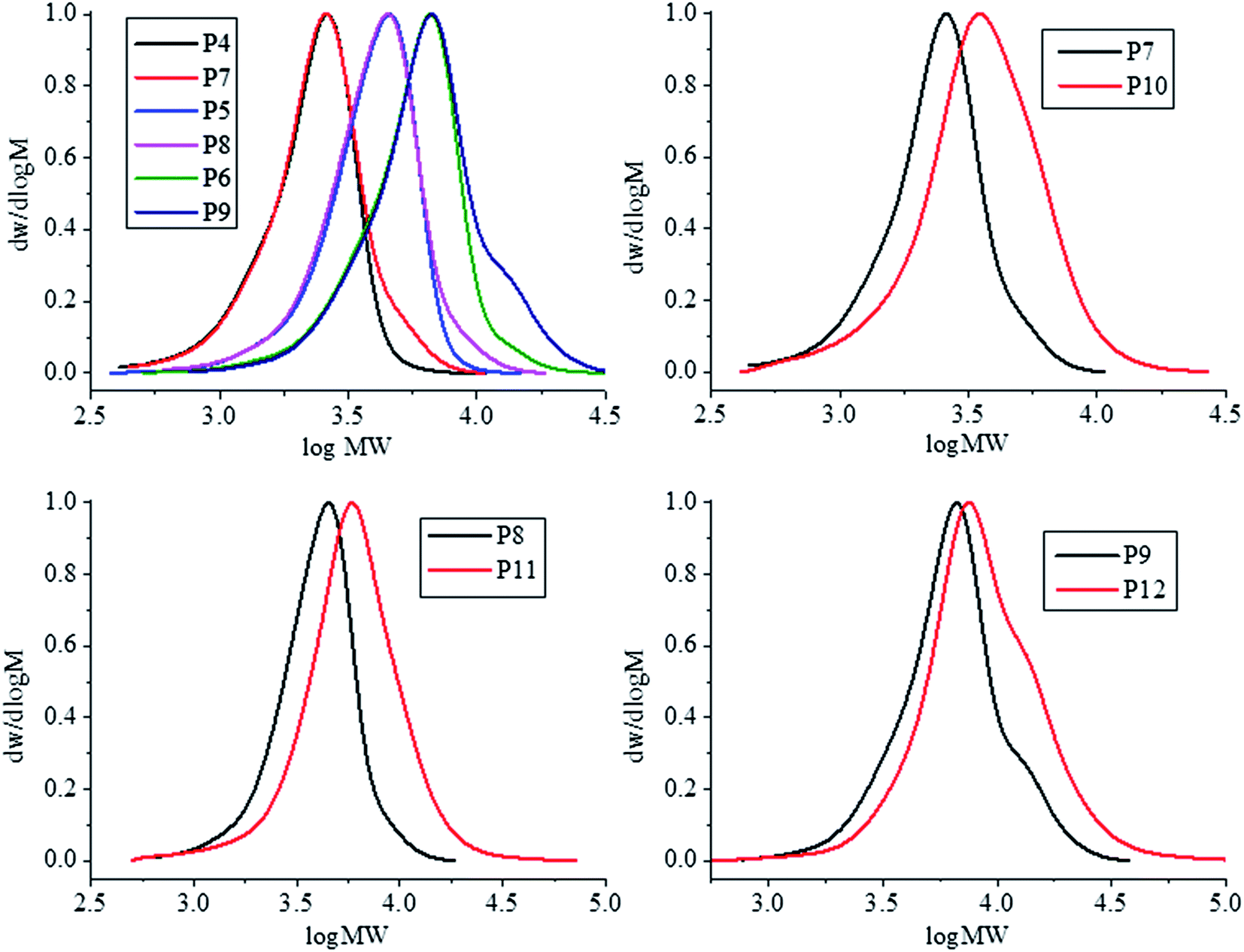 | ||
| Fig. 3 GPC traces of N3-PDMAEMA samples (P4–6) overlaid against NH2-PDMAEMA samples (P7–9) (top left) and PIB-b-PDMAEMA samples; P10 (top right), P11 (bottom left), and P12 (bottom right). | ||
Primary amine functionalised PDMAEMA was than coupled with the terminal cyclic anhydride on PIBSA to synthesise PIB-b-PDMAEMA block copolymers (Scheme 1). After 2 hours at 90 °C the reaction mixture was cooled and the resulting block copolymer was precipitated into n-hexane. Despite coupling each PDMAEMA to a hydrophobic PIB block, each block copolymer was still suitably hydrophilic and precipitated and ass such the excess PIBSA could be removed from the product. Table 3 shows the final molecular weight and dispersity for the three block copolymers synthesised (P10–12). A clear shift towards higher molecular weight, shown by the Mn and Mp values, is observed with only a slight increase in the final polymer dispersity.
| Run | NH2-PDMAEMA coupled | M n,Theo (g mol−1) | M n,GPC (g mol−1) | M p,GPC (g mol−1) | Đ |
|---|---|---|---|---|---|
| P10 | P7 | 1850 | 2900 | 3500 | 1.37 |
| P11 | P8 | 3180 | 4800 | 5800 | 1.43 |
| P12 | P9 | 4560 | 7000 | 7500 | 1.45 |
GPC chromatograms of the PIB-b-PDMAEMA overlaid against their respective H2N-PDMAEMA samples is shown in Fig. 3. Each pair of chromatograms show a shift to higher molecular weight after the coupling reaction (red line), this shift is most dramatic for the lowest molecular weight PDMAEMA coupled (P7).
These GPC traces also show the formation of a slight high molecular weight shoulder forming after coupling. It is possible for PIBSA to be doubly functionalised with two cyclic anhydride groups. These difunctionalised PIBs could therefore react with two primary amine functionalised PDMAEMA chains leading to a higher molecular weight species than the desired block copolymer. Furthermore, H2N-PDMAEMA may also react with the formed acid following reaction of H2N-PDMAEMA with PIBSA. This would also lower the purity of obtaining pure PIB-b-PDMAEMA diblocks. Reaction of H2N-PDMAEMA and the formed acid could be suppressed by utilising an excess of PIBSA to promote coupling between cyclic anhydride and primary amine as intended.
Conclusions
Two synthetic strategies for the synthesis of PIB-b-PDMAEMA from commercially available PIB have been presented. SET-LRP of DMAEMA using a PIB macroinitiator has been utilised to synthesise PIB-b-PDMAEMA copolymers, thus combining PIB macroinitiators with another controlled radical polymerisation technique. Longer block lengths of PDMAEMA could be synthesised by increasing the ratio of monomer to initiator. However, PIB-b-PDMAEMA copolymers contained residual PIB following polymerisation as the PIB macroinitiator had low end group fidelity (60%) and therefore contained a significant portion of PIB that could not be chain extended. Moreover, low end group fidelity of the PIB macroinitiator meant that targeted block lengths of PDMAEMA were challenging to synthesise as the actual monomer to initiator ratio was higher than intended. This is evidenced by measured molecular weight being much greater than the theoretical value. Fortunately the residual PIB impurity could be removed by selective precipitation of the PIB-b-PDMAEMA copolymers in n-hexane and thus furnish the desired block copolymers.An alternative synthetic pathway to PIB-b-PDMAEMA copolymers, via reactive coupling of H2N-PDMAEMA and PIBSA, was devised to not utilise the PIB macroinitiator. H2N-PDMAEMA was synthesised at different molecular weights by SET-LRP of DMAEMA using an azide functional initiator, said azide was then subsequently reduced to a primary amine post-polymerisation. Polymerising DMAEMA before coupling with PIBSA allowed for the synthesis of well-defined PDMAEMA to a targeted DP. DP could be better targeted by this approach as the monomer to initiator ratio was better defined than before when polymerising from a PIB macroinitiator with low end group fidelity. H2N-PDMAEMA and PIBSA were then coupled together at 90 °C to yield PIB-b-PDMAEMA copolymers. Residual PIB impurities could again be removed by selective precipitation of PIB-b-PDMAEMA into n-hexane. PIB-b-PDMAEMA copolymers were shown to have measured molecular weights greater than the measured molecular weight of the H2N-PDMAEMA precursors and dispersities were reasonably low considering the PIBSA provided had a dispersity of 1.87. A drawback of this coupling approach is that the presence of difunctionalised PIBSA may result in two H2N-PDMAEMA chains coupling to one PIB. This would deviate from the synthesis of pure block copolymers and is evidenced by a high molecular weight shoulder on the GPC traces of all three PIB-b-PDMAEMA samples. This drawback could be remedied by utilising PIBSA which is strictly monofunctionalised and will form the basis of future investigation for this study. We believe coupling of polymers bearing primary amine end groups and PIBSA represents a versatile new method of synthesising PIB based block copolymers.
References
- J. P. Kennedy and M. Hiza, J. Polym. Sci., Part A: Polym. Chem., 1983, 21, 1033–1044 CrossRef CAS
.
- R. Tripathy, U. Ojha and R. Faust, Macromolecules, 2009, 42, 3958–3964 CrossRef CAS
- C. P. Roche, M. R. Brei, B. Yang and R. F. Storey, ACS Macro Lett., 2014, 3, 1230–1234 CrossRef CAS
- J. P. Kennedy, S. Midha and A. Gadkari, Journal of Macromolecular Science, Part A: Chemistry, 1991, 28, 209–224 CrossRef
- S. K. Jewrajka and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2612–2623 CrossRef CAS
- G. Kali, S. Vavra, K. László and B. Iván, Macromolecules, 2013, 46, 5337–5344 CrossRef CAS
- T. K. Georgiou, C. S. Patrickios, P. W. Groh and B. Ivan, Macromolecules, 2007, 40, 2335–2343 CrossRef CAS
- G. Kali, T. K. Georgiou, B. Ivan and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4289–4301 CrossRef CAS
- W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749–1759 CrossRef CAS
- H. Barqawi, M. Schulz, A. Olubummo, V. Saurland and W. H. Binder, Macromolecules, 2013, 46, 7638–7649 CrossRef CAS
- U. Haldar, K. Bauri, R. Li, R. Faust and P. De, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1125–1133 CrossRef CAS
- K. Bauri, P. De, P. N. Shah, R. Li and R. Faust, Macromolecules, 2013, 46, 5861–5870 CrossRef CAS
- R. F. Storey and B. J. Chisholm, Polymer, 1996, 37, 2925–2938 CrossRef CAS
- T. Pernecker, J. P. Kennedy and B. Iván, Macromolecules, 1992, 25, 1642–1647 CrossRef CAS
- J. Feldthusen, B. Iván and A. H. E. Muller, Macromolecules, 1998, 31, 578–585 CrossRef CAS
- A. Takacs and R. Faust, Macromolecules, 1995, 28, 7266–7270 CrossRef CAS
- A. J. D. Magenau, N. Martinez-Castro and R. F. Storey, Macromolecules, 2009, 42, 2353–2359 CrossRef CAS
- C. Ren, X. Liu, X. Jiang, G. Sun and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1143–1150 CrossRef CAS
- X. Chen, B. Iván, J. Kops and W. Batsberg, Macromol. Rapid Commun., 1998, 19, 585–589 CAS
- Z. Fang and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3662–3678 CrossRef CAS
- Z. Fang and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3679–3691 CrossRef CAS
- Á. Szabó, A. Wacha, R. Thomann, G. Szarka, A. Bóta and B. Iván, J. Macromol. Sci., Pure Appl. Chem., 2015, 52, 252–259 CrossRef
- C. Ren, X. Jiang, G. Lu, X. Jiang and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1478–1486 CrossRef CAS
- A. Guerrero, K. Kulbaba and M. Bochmann, Macromolecules, 2007, 40, 4124–4126 CrossRef CAS
-
S. P. Srivastava and J. Hancsók, Fuels and Fuel-Additives, John Wiley & Sons, 1st edn, 2014, ch. 5, pp. 177–208 Search PubMed
-
R. E. Cherpeck and K. D. Nelson, US Pat., 5993497, 1999
- C. Hongfa, J. Tian, H. S. Bazzi and D. E. Bergbreiter, Org. Lett., 2007, 9, 3259–3261 CrossRef CAS PubMed
- D. E. Bergbreiter, C. Hobbs, J. Tian, H. Koizumi, H.-L. Su and C. Hongfa, Pure Appl. Chem., 2009, 11, 1981–1990 Search PubMed
- B. Ivan, J. P. Kennedy and V. S. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 1980, 18, 3177–3191 CrossRef
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS
- X. Jiang, S. Fleischmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753–1763 CrossRef CAS
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS
-
R. Bloch, T. J. McCrary and D. W. Brownawell, US Pat., 4517104, 1985
-
A. O. Patil, US Pat., 5439607, 1995
-
K. J. Duyck and T. E. Nalesnik, US Pat., 6916767, 2005
-
J. R. Blackborow and M. J. Clarke, US Pat., 5644001, 1997
-
P. Gateau, D. Binet and J.-P. Durand, US Pat., 5739355, 1998
-
R. E. Cherpeck, US Pat., 6352566B1, 2002
-
M. Heddadj, W. R. Ruhe and G. P. Sinquin, US Pat., 7091306B2, 2006
- C. A. Orr, J. J. Cernohous, P. Guegan, A. Hirao, H. K. Jeon and C. W. Macosko, Polymer, 2001, 42, 8171–8178 CrossRef CAS
- X. Jiang, M. C. Lok and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2077–2084 CrossRef CAS PubMed
- M. Zhang, P. A. Rupar, C. Feng, K. Lin, D. J. Lunn, A. Oliver, A. Nunns, G. R. Whittell, I. Manners and M. A. Winnik, Macromolecules, 2013, 46, 1296 CrossRef CAS
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS
- G.-Y. Shi, X.-Z. Tang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2390 CrossRef CAS
- S. Sun, Y. Cao, J. Feng and P. Wu, J. Mater. Chem., 2010, 20, 5605 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15346a |
| This journal is © The Royal Society of Chemistry 2016 |