
New chloramphenicol Schiff base ligands for the titanium-mediated asymmetric aldol reaction of α,β-unsaturated aldehydes with diketene: a short synthesis of atorvastatin calcium†
Abstract
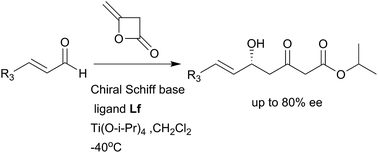
Several novel chiral Schiff base ligands were prepared from a commercially available chloramphenicol base and applied to the titanium-mediated asymmetric aldol reaction of diketene with various α,β-unsaturated aldehydes. This reaction proceeded in good yield with high enantioselectivity. The synthetic utility of this methodology was demonstrated in the short synthesis of atorvastatin calcium.
 Please wait while we load your content...
Please wait while we load your content...

