DOI:
10.1039/C6RA15267H
(Paper)
RSC Adv., 2016,
6, 92804-92812
Room-temperature fabrication of a three-dimensional reduced-graphene oxide/polypyrrole/hydroxyapatite composite scaffold for bone tissue engineering†
Received
15th June 2016
, Accepted 8th September 2016
First published on 20th September 2016
Abstract
The development of tissue engineering (TE) provides a promising alternative strategy for bone healing and regeneration. For the successful application of TE, a scaffold with a three-dimensional (3D) hierarchical structure is required to provide sites for cell adhesion and proliferation. In the present study, by using nickel foam (NF) as a template, we reported a simple but low-cost strategy to fabricate 3D reduced-graphene oxide (rGO) and 3D rGO/polypyrrole (3D rGO/PPY) struts through an electrostatic layer-by-layer assembly strategy followed by an electrochemical deposition process. The results indicated that both 3D rGO and rGO/PPY could replicate the morphology of NF, while the 3D rGO/PPY exhibits better mechanical property and can be processed into the desired configuration. After mineralization, the 3D rGO/PPY/HA scaffold can keep MC3T3-E1 cells at an active proliferation state with 6.6 times upregulation at the 4th day, superior to that of the 3D rGO/PPY scaffold (5.27 times) and control group. The excellent osteoblastic performance of the 3D rGO/PPY/HA scaffold can be attributed to the good biocompatibility of HA, and the unique 3D macro-/nano-hierarchical binary structure with high specific surface area and large surface roughness. Our finding suggests that the mineralized 3D rGO/PPY can be considered as an attractive scaffold for bone healing and regeneration in the future.
1. Introduction
The treatment of bone defects, caused by fracture, cancer resection, serious infection or osteo-degenerative diseases, imposes a tremendous socioecological burden on the patient's family and whole community.1 Traditionally, the bone defect is mainly managed by bone autograft, allograft, and xenografts.2 These methods always encounter serious drawbacks, such as an insufficient supply of graft tissue, significant donor-site morbidity, unpredictable bone resorption, long-term host immune rejection, as well as extended surgical time, and hospital stays.3–6
The emergence of bone tissue engineering technology offers a promising solution to resolve the encountering challenges and becomes a hot spot research area in orthopaedic surgery in recent few years. The bone tissue engineering mainly involves the implantation of the scaffolds alone or in combination with the growth factor, cell and gene delivery to regenerate the injured issues.7,8 Inspired by the fact that hydroxyapatite (HA) is the main component of the hard tissue of our body, considerable efforts have been devoted to exploring the promising scaffold to support hydroxyapatite or related calcium phosphates. Up to now, several types of scaffolds, including BCP,9 bioactive glass,10 poly(lactic-co-glycolic) acid (PLGA),11 PELA,12 PU13 have been successfully developed. Extensive experimental results confirmed that the scaffold-supported hydroxyapatite and/or calcium phosphate composites can more effectively improve the integration of biomaterials with bone-forming cells and further serve as a template for further growth of bone tissue, superior to that of individual hydroxyapatite and/or calcium phosphate powders.14,15 Despite the remarkable progress, the prepared scaffolds are nanofilm, nanoparticles or porous materials with uneven nanoscaled porous structure, which induce the non-uniform deposition of bioactive material within its internal nanoscaled porous structure. Therefore, there still are great difficulties in mimic the three-dimensional hierarchical macrostructure of the bone in our body.
It has been well established that, besides the excellent biocompatibility and satisfactory mechanical properties, a good scaffold should possess 3D hierarchical macro-/nano-structure for enhancing tissue regeneration.16,17 Numerous experimental results suggest that the 3D hierarchical scaffold with the large pore size of 200–500 μm and porous bioactive materials that deposited on backbone of scaffold could more effectively favor the tissue growth and capillary formation.15,16 The macro-sized pores in scaffolds benefit the transport of nutrients and simultaneously provide the large specific area for the tissue growth.18 The porous nanocomposite coating (i.e., hydroxyapatite and/or calcium phosphate composites) on the skeleton of scaffold could remarkably enhance the adhesion of osteoblast at the early stage of implantation, and stimulate the proliferation and differentiation of osteoblast at next stage of implant assimilation.19 Under this guideline, Kim and co-workers20 successfully prepared a hierarchical macro-/nano-structured polylactic-co-glycolic acid (PLGA) scaffold by a capillary force-driven lithography strategy in the combination of a macro-wrinkling method. Oh et al.21 developed a hierarchical structure on collagen scaffolds through an ice frozen-driven strategy. Unfortunately, high-temperature sintering or complex-stepped fabrication process is involved. Moreover, the scaffold fabricated through frozen-driven strategy generally deliver the macropores with size ranging from 5 to 15 μm,22 which is far from the optimized size of 200–500 μm in tissue engineering, limiting its applications in bone regeneration. Therefore, there is a great need to develop low-cost method for fabricating hierarchical macro-/nano-structured 3D scaffold with large macro-sized pores.
Graphene foam, a three-dimensional graphene porous geometry configuration with the pore size of 100–300 μm, possesses many fascination properties such as large surface area, good elasticity, and high mechanical strength,23,24 and has been extensively utilized in portable electronic devices and energy storage/conversion systems.25–27 In bone tissue engineering, 3D graphene foam also is one of promising scaffold for bone tissue engineering because of its good biocompatibility, excellent anti-corrosion performance and typical 3D topographical structure for cell proliferation/differentiation.28–31 Moreover, the cross-linked skeleton in 3D graphene foam allows us to tailor its mechanical properties and bone-bond performance by depositing polymer (e.g., polypyrrole) or bioactive materials. These advantages pave the way for the potential application of 3D graphene oxide foam in bone tissue engineering. However, currently, the synthesis of 3D graphene foam was usually performed at the high temperature (900–1000 °C) with dangerous gas as the carbon source,32–34 the yield of 3D graphene foam is also very low, limiting its potential application in bone tissue engineering.
With these in mind, in the present study, by using nickel foam (NF) as the template, we reported a simple but low-cost method to fabricate highly conductive 3D reduced-graphene oxide/poly-pyrrole (3D rGO/PPY) struts through an electrostatic layer-by-layer (LBL) assembling strategy followed by an electrochemical deposition process. All fabrication process is completed at room temperature. Due to its good mechanical properties, after NF template was removed, the prepared 3D rGO/PPY scaffold ideally replicated the framework of NF, and presented a macroporous morphology with pore size ranging from 100 to 300 μm, allowing the homogeneous deposition of bioactive material within its internal surface. With the 3D rGO/PPY strut as a matrix, we successfully fabricated the 3D hierarchical rGO/PPY/HA scaffold through an electrochemical mineralization process and investigated its application in bone regeneration. Results demonstrate that the prepared 3D hierarchical rGO/PPY/HA scaffold has strong adhesion to MC3T3-E1 osteoblast cell, and can significantly improve the proliferation and differentiation of osteoblast at the early stage of bone formation. Combination its excellent osteogenic performance, simple but low-cost fabrication process, satisfactory mechanical properties as well as desired geometry configuration shape, we believe that the prepared 3D hierarchical rGO/PPY/HA scaffold has a promising application in bone tissue engineering in coming future.
2. Experimental section
2.1 Preparation of 3D rGO/PPY scaffold
GO suspension was synthesized by oxidizing graphite according to a modified Hummer's method.35,36 The resulting GO suspension was diluted to 0.47 mg mL−1 for usage in the following experiment. Before assembly, nickel foam (NF, 20 × 20 × 1.6 mm) was sequentially cleaned with diluted hydrochloric acid, acetone and deionized water, then immersed into the 20% poly(diallyl dimethylammonium) chloride (PDDA, Mw 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000) aqueous solution for 2 h to modify the surface of NF with positively charged PDDA molecules. Subsequently, the PDDA-decorated NF was placed into the GO suspension for 2 h to absorb the GO nanosheets. Then, the NF was taken out and carefully rinsed with deionized water to wash away the physically adsorbed GO nanosheet followed by dried at 80 °C for 30 min. After repeated the above steps for 5–7 times, a compact NF/rGO foam was obtained. The deposition of PPY on NF/rGO substrate was performed on a commercial electrochemical work station (CHI 660E, Chenhua Instrument Co., Shanghai) at 0.8 V under room temperature within a three-electrode cell configuration which consists of a saturated calomel electrode as the reference electrode, a platinum plate as the counter electrode, and the prepared composite foam as the working electrode. The electrolyte containing 0.1 M pyrrole, 0.08 M lithium perchlorate and 0.112 M sodium dodecyl sulfate.37 After electrodeposition, the resulting composite foam was immersed into the hot FeCl3/HCl mixed solution to etch away the NF template followed by rinsing with deionized water for several times.
000–200000) aqueous solution for 2 h to modify the surface of NF with positively charged PDDA molecules. Subsequently, the PDDA-decorated NF was placed into the GO suspension for 2 h to absorb the GO nanosheets. Then, the NF was taken out and carefully rinsed with deionized water to wash away the physically adsorbed GO nanosheet followed by dried at 80 °C for 30 min. After repeated the above steps for 5–7 times, a compact NF/rGO foam was obtained. The deposition of PPY on NF/rGO substrate was performed on a commercial electrochemical work station (CHI 660E, Chenhua Instrument Co., Shanghai) at 0.8 V under room temperature within a three-electrode cell configuration which consists of a saturated calomel electrode as the reference electrode, a platinum plate as the counter electrode, and the prepared composite foam as the working electrode. The electrolyte containing 0.1 M pyrrole, 0.08 M lithium perchlorate and 0.112 M sodium dodecyl sulfate.37 After electrodeposition, the resulting composite foam was immersed into the hot FeCl3/HCl mixed solution to etch away the NF template followed by rinsing with deionized water for several times.
2.2 Characterization
The surface morphology, crystalline structure and physicochemical surface state of the prepared samples were characterized by field emission scanning electron microscope (FESEM, JSM-6701F, JEOL), X-ray photoelectron spectroscopy (XPS, Kratos AXIS Ultra DLD, Al Kα), X-ray powder diffraction analysis (XRD, D/max-2400, Rigaku, Cu Kα), Fourier infrared spectrometer (FT-IR, IFS66V/S, Bruker) and Raman spectrometer (HR-800, Jobin Yvon) with a 514 nm Ar-ion laser as excitation source. The mechanical properties of the prepared samples were evaluated on a TI-950 TriboIndenter (Hystron Inc., USA) equipped with a diamond tip at a maximum indentation depth of 50 nm. Before evaluation, the prepared samples was fixed on the silicon substrate and then was focused under optical microscopy to make sure that the indenter was loaded on the backbone of the 3D rGO and 3D rGO/PPY samples.
2.3 Mineralization of 3D rGO/PPY scaffold
Mineralization of 3D rGO/PPY scaffold was conducted via the mentioned electro-deposition process under −1.4 V at 30 °C for 30 min. The electrolyte was a mixed aqueous solution contains 0.042 M Ca(NO3)2, 0.1 M NaNO3 and 0.025 M NH4H2PO4.38 The pH value of the electrolyte was adjusted to 4.2 by adding diluted hydrochloric acid. After depositing, the samples were taken out from electrolyte followed by rinsing and drying at room temperature. The resulting scaffold was denoted as 3D rGO/PPY/HA.
2.4 MTT assay
The prepared 3D rGO/PPY and 3D rGO/PPY/HA scaffolds with size of 8.0 × 8.0 × 1.6 mm were sterilized under UV radiation for overnight and used for cell cultures. MC3T3-E1 osteoblast cells (a clonal mouse osteoblastic cell line, ATCC, Rockville, MD, USA) were employed to evaluate the bio-compatibility and proliferation behaviors of the prepared 3D rGO/PPY and 3D rGO/PPY/HA scaffolds. The osteoblast-like MC3T3-E1 cells were cultured at a density of 104 cells per scaffold in Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) supplemented with fetal bovine serum (FBS, 10%, Australia Origin, Gibco, USA), penicillin (100 UI mL−1), and streptomycin (100 UI mL−1) and D-glucose (4.5 g L−1) under 5% aseptic CO2 atmosphere and at 37 °C. The media were refreshed every three day until the cells reached confluence. Then, the 3D rGO/PPY and 3D rGO/PPY/HA scaffolds were placed into a 24-well plate and then seeded with 104 cell per mL concentration on each well. At a given time (1, 2 and 4 day), 100 μL of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT, St. Louis, MO, USA) solution was injected into each well. The cells were cultured for another four hours. The lower blue formazan reaction product was dissolved by adding 200 μL of dimethylsulfoxide. Then, the mixed solution was transferred into a 96-well plate, and its absorbance was measured by a microplate reader (Bio-Rad iMark) for three times and calculated the average absorbance value. For comparison, the absorbance of the cells only in the culture medium was selected as control. The proliferation performance of the as-prepared samples was obtained by comparing the absorbance of experiment group and control group (set as 100%).
2.5 Acridine orange/ethidium bromide (AO/EB) double staining
The MC3T3-E1 cells were seeded at a density of 1 × 106 cells per mL in 24-well plate and allowed to grow with 3D rGO/PPY and 3D rGO/PPY/HA scaffolds for 2, and 4 d, respectively. Then, these samples were washed with PBS for three times and dehydrated through absolute ethyl alcohol before staining with mixed AO/EB solution (100 μg mL−1 of AO and 100 μg mL−1 of EB) on a microscope slide for 1 min in the dark, and were observed immediately under fluorescence microscope (OLYMPUS BX53). The reagents AO, and EB were purchased from Sigma (St. Louis, MO, USA).
2.6 Cell adhesion and proliferation
After co-culturing for 1 and 4 days, the scaffolds (3D rGO/PPY, 3D rGO/PPY/HA) were taken out from culture medium and rinsed twice with PBS, immobilized with 2.5% glutaraldehyde at 4 °C for 2 h, dehydrated by different gradient ethanol and finally dried with supercritical CO2 drying technique. To more clearly observe the cell behavior of adhesion and proliferation under FESEM, a thin layer of Au film was coated on scaffold under vacuum to improve the electrical conductivity of the samples.
2.7 Statistical analysis
Statistical analyses were performed using SPSS version 12.0. One-way ANOVA with Duncan's multiple range tests used to examine differences between groups. The value of P < 0.05 were considered significant unless stated otherwise.
3. Results and discussion
3.1 Preparation and characterization of 3D rGO/PPY scaffold
Construction of 3D hierarchical macroporous scaffold with large pore size of 200–500 μm is essential for osteoblast differentiation and bone regeneration.16,17 In this study, by using commercial macroporous NF as template, 3D rGO/PPY scaffold was fabricated through a LBL strategy followed by an electrochemical deposition process. The detailed synthesis protocol is illustrated in Fig. 1. According to the previous report, nickel substrate has a negative zeta potential at the pH value of 3.5–9 due to the formation of NiO(OH) species on the colloid surface.39 This finding allows us to alter the chemical state of the NF by adsorbing the molecules with positive charged NH+ group in PDDA molecules through electrostatic interaction. After decorated with PDDA molecules, the surface chemical state of NF was changed from negative to positive. The positive chemical state of NF makes it easily absorb the GO nanosheets via the similar electrostatic interaction between the NH+ group in PDDA molecules and the negative functional groups (such as epoxy, carboxyl, and hydroxyl groups) on the basal plane of GO nanosheets.40 As the GO covered NF has immersed again into PDDA solution to positively charge the surface of the substrate, the color of GO nanosheets changes from light yellow to black, indicating that the graphene oxide was simultaneously reduced to graphene (named as reduced GO, rGO). The reduction of GO to rGO during the assembling process was subsequently confirmed by the XPS results (Fig. 3). By alternately repeating the above mentioned processes for several times, the GO nanosheets can be successfully assembled on the surface of NF. It should be noted that the mechanical properties of 3D graphene foam still can't be satisfied with the high requirement as an individual scaffold for bone regeneration. To further enhance its mechanical properties, we deposit an ultra-thin layer of PPY to consolidate the 3D reduced-graphene oxide foam. After etched away the NF template in the mixed solution of hot FeCl3/HCl, the 3D rGO/PPY foam was obtained.
 |
| | Fig. 1 Preparation of the 3D rGO/PPY scaffold through an electrostatic LBL strategy followed by an electrochemical deposition process. | |
The chemical state of GO and PPY film assembled on NF was characterized by FT-IR, Raman and XPS. Fig. 2(a) shows the FT-IR spectra of the 3D rGO and 3D rGO/PPY films in the range of 4000–500 cm−1. For assembled GO film, two strong peaks at 3420 and 1631 cm−1 can be attributed to O–H stretching vibration and strong carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration, respectively,40 demonstrating the presence of oxygen-containing functionalities in assembled rGO film. The peaks at ca. 1455 and 1119 cm−1 can be assigned to the carboxyl C–O stretching vibration and the epoxy C–O stretching vibration, respectively.41 Compared with the FT-IR spectrum of the GO, several new, characteristic PPY peaks can be found in rGO/PPY scaffold. The peaks at 1457, 1384, and 1042 cm−1 can be ascribed to the vibrations of –CC, –C–H, –N–H of PPY, respectively,42 suggesting that PPY was successfully anchored to the surface of the 3D rGO/PPY scaffold. The existence of PPY coating layer can be further supported by Raman spectroscopy (Fig. 2(b)). It can be seen that the original 3D rGO shows two typical bands at around 1589 cm−1 (G band) and 1348 cm−1 (D band), which can be attributed to the zone centre phonons of the E2g symmetry and K-point phonons A1g symmetry of the graphitic materials, respectively.43 For the 3D rGO/PPY scaffold, apart from the representative G and D bands of the 3D rGO, its Raman spectrum also contains several weak and wide peaks consistent with that of the PPY. Noticeably, the band at ca. 1593.5 cm−1 is weaker and shifted to lower wavenumber (from 1589.5 cm−1).44 The ID/IG ratio for 3D rGO and 3D rGO/PPY scaffold was calculated to be 1.01 and 0.92, respectively, suggesting a more disordered or defective structure of the 3D rGO/PPY scaffold.
O) stretching vibration, respectively,40 demonstrating the presence of oxygen-containing functionalities in assembled rGO film. The peaks at ca. 1455 and 1119 cm−1 can be assigned to the carboxyl C–O stretching vibration and the epoxy C–O stretching vibration, respectively.41 Compared with the FT-IR spectrum of the GO, several new, characteristic PPY peaks can be found in rGO/PPY scaffold. The peaks at 1457, 1384, and 1042 cm−1 can be ascribed to the vibrations of –CC, –C–H, –N–H of PPY, respectively,42 suggesting that PPY was successfully anchored to the surface of the 3D rGO/PPY scaffold. The existence of PPY coating layer can be further supported by Raman spectroscopy (Fig. 2(b)). It can be seen that the original 3D rGO shows two typical bands at around 1589 cm−1 (G band) and 1348 cm−1 (D band), which can be attributed to the zone centre phonons of the E2g symmetry and K-point phonons A1g symmetry of the graphitic materials, respectively.43 For the 3D rGO/PPY scaffold, apart from the representative G and D bands of the 3D rGO, its Raman spectrum also contains several weak and wide peaks consistent with that of the PPY. Noticeably, the band at ca. 1593.5 cm−1 is weaker and shifted to lower wavenumber (from 1589.5 cm−1).44 The ID/IG ratio for 3D rGO and 3D rGO/PPY scaffold was calculated to be 1.01 and 0.92, respectively, suggesting a more disordered or defective structure of the 3D rGO/PPY scaffold.
 |
| | Fig. 2 The FT-IR spectra (a) and Raman spectra (b) of the 3D rGO and 3D rGO/PPY scaffolds, respectively. | |
The chemical composition of the samples was further investigated by XPS technique. A full-survey-scan spectrum in Fig. 3(a) suggests that, except for C, N, and O elements, no other elements can be detected in the 3D rGO/PPY composite scaffold. The C 1s spectra of the as-prepared GO powder (Fig. 3(b)) displays four peaks centered at 284.5, 286.5, 287.9, and 288.9 eV, corresponding to CC/C–C, C–O, carbonyl (CO), and carboxylate (O–CO), respectively.45 After electrostatic LBL had assembled on PDDA decorated NF, the main oxygen-containing functional groups (CO, O–CO) of GO nanosheets were partially disappeared. Instead, a new fitted peak (C–N) at 285.4 eV appeared (Fig. 3(c)), which can be attributed to the formation of C–N bond between the NH+ group of PDDA molecules and functional groups on graphene oxide sheets.46 This reveals that the GO were partially reduced to rGO during the electrostatic LBL assembly process. After PPY coating, the detailed chemical state of PPY was characterized by the N 1s spectra. Fig. 3(d) shows the N 1s spectra of 3D rGO/PPY composite scaffold. In the N 1s spectrum, three peaks centered at 397.6, 399.5, and 400.8 eV can be found, which correspond to imine nitrogen (–N), N–H bond, and positively charged nitrogen (–N+), respectively.47 Based on the above results, it can be concluded that PPY layer was successfully deposited on rGO matrix.
 |
| | Fig. 3 (a) XPS survey spectrum of the 3D rGO and 3D rGO/PPY scaffolds, (b) C 1s spectrum of GO powder before LBL process, (c) C 1s spectrum of 3D rGO scaffold after LBL assembly and (d) N 1s spectra of the 3D rGO/PPY scaffold. | |
Morphological and structural characterization of the as-prepared 3D rGO and 3D rGO/PPY foam were performed by FESEM, XRD, and Raman microspectrometer. Fig. 4(a) shows the low-magnification FESEM image of NF after assembling the GO nanosheets for five times. As can be seen that the graphene nanosheet tightly adhered on the skeleton of NF, and no rGO floccules are detected on the backbone of NF template. The as-deposited rGO film on NF can be easily detected by XRD and Raman microspectrometer (Fig. S1† and Fig. 2(b)). High-magnification FESEM image, as shown in Fig. 4(b), demonstrates that rGO film with some negligible bubbles and ripples are smoothly spread along the NF surface. No obvious crakes and defects can be detected near the grain boundary of NF, suggesting that the rGO film can be deposited on the skeleton of the macroporous matrix without the limitation of the gain boundary of NF. The surface morphology of as-assembled rGO film under room temperature is roughly similar to that of graphene film grown under high temperature using NF as the template.32,48 The compact stack of GO film on NF is closely related to the strong electrostatic interaction between NH+ group in PDDA molecules and negatively charged functional groups on the basal plane of GO nanosheets as well as the strong π–π interaction between GO nano-sheets. Inspired by the good environmental stability, high conductivity, excellent flexibility and facile synthesis of PPY, we deposit a thin layer of PPY on NF/rGO matrix via an electrochemical strategy to further consolidate the mechanical property of 3D rGO foam. As shown in Fig. 4(c) and (d) after electrochemical deposition, the PPY film consisting interconnected nanoparticles with size ranging from 150–300 nm is uniformly deposited on the backbone of NF/rGO foam, forming a typical foam-like 3D NF/rGO/PPY composite. It also can be observed that the deposition of PPY greatly increase the surface roughness of the skeleton of the NF/rGO, which would favor the construction of nanostructure on the backbone of NF/rGO via an extra mineralization strategy and thus expand its application in tissue engineering. Surprisingly, after NF template was completely removed (Fig. 4(e) and Fig. S1†), the rGO/PPY composite remains the 3D cross-linked network structure of NF and demonstrate a 3D self-supported full-carbon foam. Higher-magnification FESEM, as shown in Fig. 4(f), suggests that the 3D rGO/PPY foam perfectly replica the surface morphology of NF with identical pore size and backbone width. More importantly, the involved room-temperature electrostatic LBL assembly strategy for construction of 3D reduced graphene foam can be realized in most chemistry/material laboratories without using any special and expensive equipment, superior to the reported strategy in previous studies, such as the freeze-drying,49 the high temperature sintering process,50 and the electro-spinning route.51
 |
| | Fig. 4 SEM micrographs of the (a and b) 3D NF/rGO, (c and d) 3D NF/rGO/PPY, and (e and f) 3D rGO/PPY scaffolds. | |
It is of interest to note that arbitrary configuration, such as helical, O-ring, and U-shaped rGO/PPY film, also can be easily fabricated by using the corresponding shape of NF as the template (Fig. 5), implying that the configuration of 3D rGO/PPY full-carbon foam only depends on the shape of NF template. This allows us to precisely design the preferred configuration of rGO/PPY scaffold according to the requirement of bone defects without complicated subsequent reprocessing.
 |
| | Fig. 5 Optical image of the prepared 3D rGO/PPY scaffold with (a) helical, (b) O-ring, and (c) U-shaped configuration. | |
3.2 Mechanical properties of 3D rGO and 3D rGO/PPY scaffold
To quantitatively analyze the effect of PPY film on the mechanical properties of 3D rGO scaffold, we measured the hardness and Young's modulus of the prepared 3D rGO and 3D rGO/PPY scaffolds by using indentation technique. Fig. 6(a) illustrates the load–displacement curves of two scaffolds during the loading/unloading process of the diamond indenter. Obviously, the scaffold of 3D rGO/PPY is more resistant to indenter and higher load than 3D rGO scaffold when the indenter tip reaches the same depth. According to their loading–displacement curves, the hardness and Young's modulus of 3D rGO and 3D rGO/PPY are calculated and illustrated in Fig. 6(b). As demonstrated, the hardness (92.27 ± 4.03 MPa) and Young's modulus (185.94 ± 10.76 MPa) of 3D rGO/PPY scaffold were almost twice as high as the corresponding values of 3D rGO with the hardness of 48.59 ± 4.96 MPa and Young's modulus of 91.0 ± 4.19 MPa. The Young's modulus of 3D rGO/PPY scaffold is also higher than that of nature cancellous bone of mandible (114 ± 78 MPa) in the infero-superior direction,52 well meeting the requirements of clinical operation for repairing of trabeculae defects.
 |
| | Fig. 6 (a) Load–displacement curves and (b) hardness/Young's modulus of the 3D rGO and 3D rGO/PPY scaffolds. | |
3.3 Mineralization of 3D rGO/PPY scaffold
Biomimetic mineralization is an effective way to endow the substrate with good biocompatibility and osteointegration ability.45
In this study, by using the electrochemical method, we deposit the biocompatible HA layer on the as-prepared 3D rGO/PPY matrix. Fig. 7 and 8 show the surface morphology and crystalline structure of the deposited HA layer under −1.4 V and 30 °C for 30 min. As shown in Fig. 8, the diffraction peaks of the depositing layer match well with those of HA (JCPDS no. 09-0432).53 The Ca/P molar ratio is calculated to be 1.62, which is much close to that in pure HA (Fig. S2 and Table S1†). The XRD and XPS results together confirm the successful deposition of HA layer on 3D rGO/PPY foam. Low-magnification FESEM image demonstrates that HA coating layer is uniformly deposited on the backbone of the 3D rGO/PPY. No serious HA aggregations were clearly observed (Fig. 7(a)). High-magnification FESEM indicates that the HA coating consists numerous porous HA nanosphere with the size of 50–100 nm. The deposition of nanosized HA spheres on the backbone of the macro-porous rGO/PPY produced a typical macro-/nano-hierarchical structure with enhanced surface roughness and surface specific surface area, which were believed to facilitate the cell adhesion and proliferation.38
 |
| | Fig. 7 SEM micrographs of 3D rGO/PPY scaffold after mineralization by electro-chemical deposition under −1.4 V@30 °C for 30 min: (a) low-magnification and (b) high-magnification. | |
 |
| | Fig. 8 XRD patterns of the prepared 3D NF/rGO/PPY, 3D rGO/PPY, 3D rGO/PPY/HA scaffold and the standard data of HA. | |
3.4 MTT assay
The viability and proliferation rate of MC3T3-E1 cells on 3D rGO/PPY and 3D rGO/PPY/HA scaffolds were assessed by using the MTT assay. As shown in Fig. 9, at 1st culture day, the proliferation rate of MC3T3-E1 cells on two scaffolds are comparable to that of the control sample. At 2nd culture day, the cell number was greatly increased, indicating the non-cytotoxicity of two prepared scaffolds. Surprisingly, as the culture period increased up to 4 days, the proliferation rate of MC3T3-E1 cell on 3D rGO/PPY and 3D rGO/PPY/HA is tremendously increased and reaches as high as 5.27 and 6.6 times, respectively, as compared to that of the control group. Moreover, in all culture period except for 1 day, the cell number on 3D rGO/PPY/HA scaffold is always higher than that on 3D rGO/PPY scaffold, suggesting the good biocompatibility of HA layer and its significant role in boosting the cell proliferation.
 |
| | Fig. 9 Comparison proliferation rate of MC3T3-E1 cells after being cultured with 3D rGO/PPY and 3D rGO/PPY/HA scaffold at 1, 2, and 4 days. Asterisk (*) indicates the significance with P < 0.05, and the error bar represents the standard deviation. | |
3.5 AO/EB double staining and fluorescence microscopy
The cytostatic activity of MC3T3-E1 cells on the as-prepared scaffolds were determined by AO/EB double staining under fluorescence microscopy. It has been established that only the AO is permeable to live cells and fluoresce green in color, while only EB is permeable to dead cells and fluoresce orange-red in color.54,55 It can be seen from Fig. 10, at 2nd and 4th culture day, the fluorescence images of MC3T3-E1 cells on two scaffolds presents as green color rather than the red color (the necrosis cells), indicating the good health of the cells on two scaffolds. Further observations suggest that the cell volume and cell number on two scaffolds at 4th culture day (Fig. 10(c) and (d)) are much larger than those at 2nd culture day (Fig. 10(a) and (b)). At 4th culture day, certain amount of filopodias is easily observed to extend from the cell bodies. In addition, at 4th culture day, the cell number on 3D rGO/PPY/HA scaffold is also higher than that on 3D rGO/PPY, these findings are consistent with the MTT results, thereby further confirming the non-cytotoxicity of two scaffolds to osteoblasts and the better proliferation ability of the 3D rGO/PPY/HA over the 3D rGO/PPY scaffold.
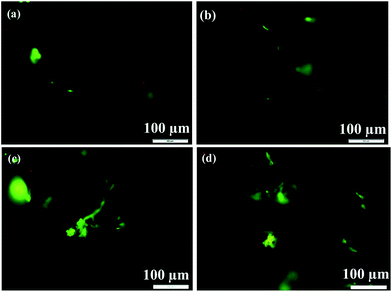 |
| | Fig. 10 Fluorescence microscope images of MC3T3-E1 cells after AO/EB double staining on 3D rGO/PPY (left alignment) and 3D rGO/PPY/HA scaffolds (right alignment) for 2, and 4 d, respectively. | |
3.6 Cell adhesion on the 3D rGO/PPY and 3D rGO/PPY/HA scaffold
The adhesion ability of MC3T3 cells after being cultured on 3D rGO/PPY and 3D rGO/PPY/HA scaffolds were observed by FESEM (Fig. 11). As shown in Fig. 11(a), after being cultured for 1 day, the cell on 3D rGO scaffold shows the typical hemispherical shape and protrudes out from the backbone of the 3D rGO/PPY scaffold. As the culture period increases up to 4 days, no obvious changes in the morphology of the cell besides the increasing of the cell size. Contrastively, at 1st culture day, the cell on rGO/PPY/HA scaffold demonstrates obvious fusiform-like structure with microscopic-filopodia at the edge area, much different from that of 3D rGO/PPY scaffold at the same culture period. According to the previous result, the existing of filopodia at the edge of cell would make it tightly bind to the surface of matrix and thus greatly improve the adhesion between cell and scaffold.56 At the 4th day, the volume of the cells on 3D rGO/PPY/HA scaffold greatly increases and spreads over a larger area on matrix (see the circled area in Fig. 11(d)). Moreover, the pseudopods at the edge of the cells also notably increased. By summarizing the surface morphology and viability of cells on two prepared scaffolds, it can be concluded that the either the cell viability or the morphology on 3D rGO/PPY/HA is superior to that on 3D rGO/PPY scaffold. The studies reported previously have indicated that the induction of HA coating can enhance the specific area,57 roughness,58 and interfacial hydrophilicity.59 These properties mentioned above all benefit for cell adhesion, proliferation, and differentiation. Moreover, the HA itself can improve the osteointegration for dental and orthopaedic implants because of its similarity in chemistry and structure to the inorganic component of natural bone.60
 |
| | Fig. 11 FESEM images of the MC3T3 cells after being cultured on 3D rGO/PPY scaffold (a and c) and 3D rGO/PPY/HA scaffold (b and d) at 1 and 4 day, respectively. | |
4. Conclusions
We have successfully fabricated the two highly conductive 3D rGO and 3D rGO/PPY foam with large macropore-size through an electrostatic LBL assembly strategy followed by an electrochemical deposition process. As compared with that of 3D rGO, the prepared 3D rGO/PPY foam demonstrated better mechanical property and thus can be processed into the desired configuration. After depositing HA mineral layer, both the surface roughness, specific surface area and the biocompatibility of the 3D foam were greatly improved. When tested with MC3T3-E1 cell, the prepared 3D rGO/PPY/HA scaffold show excellent cell proliferation capability and proliferation state with 6.6 times of upregulation at 4th culture days, much superior to that of the 3D rGO/PPY scaffold and control sample, suggesting its promising applications in bone healing and regeneration. More importantly, we provide a simple but low-cost and room-temperature method for preparing the 3D foam-like struts with the desired configuration, which would significantly facilitate its fast clinical applications in tissue engineering.
Acknowledgements
We thank the Natural Science Foundation of China (81571829), the Fundamental Research Funds for the Central Universities (lzujbky-2015-35, lzujbky-2015-289), and the Fundamental Research Funds for the Central Universities under Grant No. lzujbky-2015-294.
References
- G. Chen, L. Yang and Y. Lv, J. Biomed. Mater. Res., Part A, 2016, 104, 833–841 CrossRef CAS PubMed.
- T. A. van Vugt, J. Geurts and J. J. Arts, BioMed Res. Int., 2016, 2016, 6984656 CAS.
- L. Ohayon, Int. J. Periodontics Restorative Dent., 2011, 31, 237–245 Search PubMed.
- J. Hao, A. Acharya, K. Chen, J. Chou, S. Kasugai and N. P. Lang, Clin. Oral Implants Res., 2015, 26, 1–7 CrossRef CAS PubMed.
- I. K. Lee, H. C. Lim, J. S. Lee, J. Y. Hong, S. H. Choi and U. W. Jung, Clin. Oral Implants Res., 2016, 27, 622–628 Search PubMed.
- S. A. J. Kronig, R. J. G. van der Mooren, E. M. Strabbing, L. H. M. Stam, J. A. S. L. Tan and E. de Jongh, et al. , Int. J. Oral Surg., 2016, 45, 507–512 Search PubMed.
- A. Y. Gamal, K. A. Abdel-Ghaffar and V. J. Iacono, J. Periodontal Res., 2016, 51, 407–416 CrossRef CAS PubMed.
- P. Bhattacharjee, D. Naskar, T. K. Maiti, D. Bhattacharya and S. C. Kundu, J. Colloid Interface Sci., 2016, 472, 16–33 Search PubMed.
- M.-H. Lee, C. You and K.-H. Kim, Materials, 2015, 8, 1150–1161 CrossRef CAS PubMed.
- X. Wang and W. Li, Nanotechnology, 2016, 27, 225102 CrossRef PubMed.
- T. Ren, J. Ren, X. Jia and K. Pan, J. Biomed. Mater. Res., Part A, 2005, 74, 562–569 CrossRef PubMed.
- A. B. Kutikov, J. D. Skelly, D. C. Ayers and J. Song, ACS Appl. Mater. Interfaces, 2015, 7, 4890–4901 CrossRef CAS PubMed.
- G. Tetteh, A. S. Khan, R. M. Delaine-Smith, G. C. Reilly and I. U. Rehman, J. Mech. Behav. Biomed. Mater., 2014, 39, 95–110 Search PubMed.
- Z. S. Tao, W. S. Zhou, K. K. Tu, Z. L. Huang, Q. Zhou and T. Sun, et al. , Injury, 2015, 46, 2164–2169 Search PubMed.
- F. Y. Teng, W. C. Chen, Y. L. Wang, C. C. Hung and C. C. Tseng, Bioinorg. Chem. Appl., 2016, 2016, 3837679 Search PubMed.
- G. S. Lee, J. H. Park, U. S. Shin and H. W. Kim, Acta Biomater., 2011, 7, 3178–3186 Search PubMed.
- T. A. Martin, S. R. Caliari, P. D. Williford, B. A. Harley and R. C. Bailey, Biomaterials, 2011, 32, 3949–3957 Search PubMed.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- R. Dua, J. Centeno and S. Ramaswamy, J. Biomed. Mater. Res., Part B, 2014, 102, 922–932 Search PubMed.
- J. Kim, W. G. Bae, H. W. Choung, K. T. Lim, H. Seonwoo and H. E. Jeong, et al. , Biomaterials, 2014, 35, 9058–9067 Search PubMed.
- H. H. Oh, Y. G. Ko, H. Lu, N. Kawazoe and G. Chen, Adv. Mater., 2012, 24, 4311–4316 CrossRef CAS PubMed.
- Y. E. Shin, Y. J. Sa, S. Park, J. Lee, K. H. Shin and S. H. Joo, et al. , Nanoscale, 2014, 6, 9734–9741 RSC.
- M. Mecklenburg, A. Schuchardt, Y. K. Mishra, S. Kaps, R. Adelung and A. Lotnyk, et al. , Adv. Mater., 2012, 24, 3486–3490 CrossRef CAS PubMed.
- S. Murali, J. R. Potts, S. Stoller, J. Park, M. D. Stoller and L. L. Zhang, et al. , Carbon, 2012, 50, 3482–3485 CrossRef CAS.
- J.-C. Yoon, J.-S. Lee, S.-I. Kim, K.-H. Kim and J.-H. Jang, Sci. Rep., 2013, 3, 1788 Search PubMed.
- J. Chen, K. Sheng, P. Luo, C. Li and G. Shi, Adv. Mater., 2012, 24, 4569–4573 CrossRef CAS PubMed.
- Y. R. Jeong, H. Park, S. W. Jin, S. Y. Hong, S.-S. Lee and J. S. Ha, Adv. Funct. Mater., 2015, 25, 4228–4236 CrossRef CAS.
- J. Ruan, X. Wang, Z. Yu, Z. Wang, Q. Xie and D. Zhang, et al. , Adv. Funct. Mater., 2016, 26, 1085–1097 Search PubMed.
- H. Xie, T. Cao, J. V. Gomes, A. H. Castro Neto and V. Rosa, Carbon, 2015, 93, 266–275 CrossRef CAS.
- A. Nieto, R. Dua, C. Zhang, B. Boesl, S. Ramaswamy and A. Agarwal, Adv. Funct. Mater., 2015, 25, 3916–3924 CrossRef CAS.
- S. Kang, J. B. Park, T.-J. Lee, S. Ryu, S. H. Bhang and W.-G. La, et al. , Carbon, 2015, 83, 162–172 CrossRef CAS.
- S. W. Crowder, D. Prasai, R. Rath, D. A. Balikov, H. Bae and K. I. Bolotin, et al. , Nanoscale, 2013, 5, 4171–4176 RSC.
- W. Jiang, H. Xin and W. Li, Mater. Lett., 2016, 162, 105–109 Search PubMed.
- M. Chen, S. Duan, L. Zhang, Z. Wang and C. Li, Chem. Commun., 2015, 51, 3169–3172 RSC.
- C. Wu, F. Li, Y. Zhang and T. Guo, Carbon, 2012, 50, 3622–3626 Search PubMed.
- X. Li, B. K. Tay, J. Li, D. Tan, C. W. Tan and K. Liang, Nanoscale Res. Lett., 2012, 7, 205–213 Search PubMed.
- X. Feng, Y. Shi and S. Jin, Appl. Surf. Sci., 2015, 353, 788–792 Search PubMed.
- S. Nardecchia, M. C. Serrano, M. C. Gutiérrez, M. T. Portolés, M. L. Ferrer and F. del Monte, Adv. Funct. Mater., 2012, 22, 4411–4420 Search PubMed.
- N. Hernandez, R. Moreno and A. J. Sanchez-Herencia, J. Phys. Chem. B, 2005, 109, 4470–4474 Search PubMed.
- M. Zuldesmi, A. Waki, K. Kuroda and M. Okido, Mater. Sci. Eng., C, 2015, 49, 430–435 CrossRef CAS PubMed.
- Y. Zhou, X.-C. Hu, Q. Fan and H.-R. Wen, J. Mater. Chem. A, 2016, 4, 4587–4591 RSC.
- X. She, P. Sun, X. Yu, Q. Zhang, Y. Wu and L. Li, et al. , J. Inorg. Organomet. Polym. Mater., 2014, 24, 884–889 CrossRef CAS.
- Y. Jiang, C. Hu, H. Cheng, C. Li, T. Xu and Y. Zhao, et al. , ACS Nano, 2016, 10, 4735–4741 CrossRef CAS PubMed.
- H. Kang, A. Kulkarni, S. Stankovich, R. S. Ruoff and S. Baik, Carbon, 2009, 47, 1520–1525 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller and R. D. Piner, et al. , Carbon, 2009, 47, 145–152 CrossRef CAS.
- W. Shuangyin, Y. Dingshan and D. Liming, ACS Nano, 2011, 5, 6202–6209 CrossRef PubMed.
- H. Zhou, G. Han, Y. Xiao, Y. Chang and H.-J. Zhai, J. Power Sources, 2014, 263, 259–267 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- H. Yu, D. Ye, T. Butburee, L. Wang and M. Dargusch, ACS Appl. Mater. Interfaces, 2016, 8, 2505–2510 CrossRef CAS PubMed.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li and C. Yu, et al. , Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS PubMed.
- Y. Luo, H. Shen, Y. Fang, Y. Cao, J. Huang and M. Zhang, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 6331–6339 CrossRef CAS PubMed.
- T. Cox, M. W. Kohn and T. Impelluso, J. Oral Maxillofac. Surg., 2003, 61, 481–487 CrossRef PubMed.
- C. He, F. Zhang, L. Cao, W. Feng, K. Qiu and Y. Zhang, et al. , J. Mater. Chem., 2012, 22, 2111–2119 RSC.
- V. SimiĆ, S. KolareviĆ, I. BrČEski, D. JeremiĆ and B. VukoviĆ-GaČIĆ, Turk. J. Biol., 2016, 40, 661–669 CrossRef.
- B. Taghizadeh, L. Ghavami, A. Nikoofar and B. Goliaei, Breast Cancer, 2015, 22, 382–390 Search PubMed.
- N. I. Aminuddin, R. Ahmad, H. M. Ansari, N. M. Zain, S. A. Akbar and B. Pingguan-Murphy, Mater. Des., 2016, 94, 274–279 CrossRef CAS.
- R. Cholas, S. Kunjalukkal Padmanabhan, F. Gervaso, G. Udayan, G. Monaco and A. Sannino, et al. , Mater. Sci. Eng., C, 2016, 63, 499–505 CrossRef CAS PubMed.
- W. Zhe, C. Dong, Y. Sefei, Z. Dawei, X. Kui and L. Xiaogang, Appl. Surf. Sci., 2016, 378, 496–503 CrossRef CAS.
- F. Liu, X. Jiang, S. Bao, R. Wang, B. Sun and M. Zhu, Mater. Sci. Eng., C, 2015, 53, 150–155 CrossRef CAS PubMed.
- E. Damien, K. Hing and S. Saeed, J. Biomed. Mater. Res., Part A, 2001, 66, 241–246 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD patterns of the 3D rGO/PPY scaffold and XPS spectra of the electrodeposited HA coating on the 3D rGO/PPY scaffold. See DOI: 10.1039/c6ra15267h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 










