
Diglycidylether of iso-eugenol: a suitable lignin-derived synthon for epoxy thermoset applications
Abstract
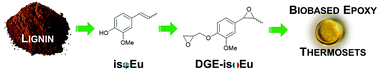
A novel lignin-based synthon, diglycidylether of iso-eugenol (DGE-isoEu) is used as a prepolymer for the preparation of thermosetting resins. DGE-isoEu is synthesized in a two-step procedure with a satisfactory yield from bio-based iso-eugenol (isoEu, 2-methoxy-4-(1-propenyl)phenol) catalytically fragmented from lignin in an organosolv process. DGE-isoEu was fully characterized by NMR, MS and FTIR. Curing of the DGE-isoEu monomer has then been investigated in the presence of several carboxylic acid derivatives hardeners. The thermal and mechanical properties of each material were recorded showing, in particular, a high Tg and instantaneous modulus values in the range of 78–120 °C and 4.6–5.5 GPa, respectively. The lignin derived new materials give very attractive thermo-mechanical properties comparable to that of common BPA-containing epoxy resins.
 Please wait while we load your content...
Please wait while we load your content...

