
Molecular-structure control of electron transfer dynamics of push–pull porphyrins as sensitizers for NiO based dye sensitized solar cells†
Abstract
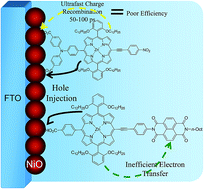
Porphyrin dyes were synthesized for use in p-type (NiO) dye sensitized solar cells based on different design principles. One porphyrin was designed with a significant charge transfer character in the excited state because of push–pull effects of the substituents. Another porphyrin had instead an appended NDI acceptor group (NDI = naphthalene diimide). The dyes were characterized by spectroscopic, electrochemical and DFT methods. Solar cells based on sensitized, meso-porous NiO showed rather poor performance compared to other organic dyes, but with a clear improvement for the dye with the NDI acceptor. Ultrafast transient absorption spectroscopy and nanosecond laser photolysis showed that hole injection into NiO was followed by unusually rapid charge recombination, predominantly on a 50–100 ps time scale, which is likely the main reason for the poor photovoltaic performance. Again the porphyrin with the NDI group showed a more long-lived charge separation that should lead to better dye regeneration in a solar cell, which can explain its better photovoltaic performance.
 Please wait while we load your content...
Please wait while we load your content...

