
The superior performance of a Pt catalyst supported on nanoporous SiC–C composites for liquid-phase selective hydrogenation of cinnamaldehyde†
Ruihua Yao,
Junrui Li,
Peng Wu and
Xiaohong Li*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663 North Zhongshan Rd., Shanghai 200062, China. E-mail: xhli@chem.ecnu.edu.cn; Fax: +86-21-62238590; Tel: +86-21-62238590
First published on 22nd August 2016
Abstract
The monometallic Pt catalyst supported on nanoporous composites containing β-SiC and amorphous carbon was proved to be active and selective for liquid-phase selective hydrogenation of cinnamaldehyde (CAL) even at room temperature. A TOF of higher than 2400 h−1 and 80% selectivity to cinnamyl alcohol (COL) were furnished by the Pt/SiC–C catalyst at 25 °C. Moreover, the Pt/SiC–C nanocatalyst showed superior performance compared to other Pt catalysts supported on carbon, silica or alumina materials under the same conditions for the tested reaction. Of particular note is that the Pt/SiC–C catalyst can be easily recovered and reused for at least 10 runs without any loss in activity or selectivity. We deduce that there is a strong interaction between Pt nanoparticles and SiC–C composites; so that there are still quite a few Pt species with positive charges on the catalyst surface even after being reduced in an aqueous solution of sodium formate at reflux temperature and pre-treated under a hydrogen flow at 400 °C, which would be helpful for preferential adsorption and activation of CAL via electrostatic interaction with the oxygen atoms of the carbonyl group of CAL, and thus COL was the primary product obtained with the Pt/SiC–C catalyst.
Introduction
Silicon carbide (SiC) has attracted much more attention in optical and electrical fields recently as one of the most important semiconductors, due to its wide bandgap, high strength, high thermal conductivity, good thermal shock resistance, low thermal expansion and good chemical inertness.1,2 Indeed, the SiC nanomaterial has been a rising star, as demonstrated by an increasing number of published research about it.3 SiC nanostructures have been successfully used as a photo-catalyst4–6 and electro-catalyst7–13 owing to their specific properties. In addition, they have been reported as an efficient support or direct metal-free catalyst for various reactions during the last decades.14–18 Recently, the thermally conductive SiC, pure or doped with different promoters, has been confirmed to be active, selective and reusable for the Fisher–Tropsch reaction.14,15 In addition, silicon carbide was also applied as a metal-free catalyst for dehydrogenation of ethylbenzene to styrene.16,17In general, silicon carbide was usually involved in some strongly exothermic gas–solid phase reactions due to its high thermal conductivity. However, the relevant researches about the utilization of SiC nanostructures as support for traditional liquid-phase catalysis are still very limited.19,20 Sometimes, silicon carbide with low specific surface area would restrict the dispersion of active sites and hinders its application in heterogeneous catalysis to some extent. Therefore, nanoporous SiC–C composites not only can reserve the special property of SiC itself, but also have large specific surface area to disperse active sites.
Herein, we developed a monometallic Pt catalyst supported on nanoporous SiC–C composites with large specific surface area for liquid-phase selective hydrogenation of CAL (Scheme 1). As well known, the selective hydrogenation of α,β-unsaturated aldehydes is challengeable in that the formation of desirable α,β-unsaturated alcohol is not thermodynamically preferred due to higher C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond energy than that of CC double bond.21 With regards of the selective hydrogenation of CAL, special supports such as graphene or carbon nanotubes for loading monometallic Pt catalyst21–27 or bimetallic catalysts (such as Pt–Fe)21,28,29 are usually required to achieve high selectivity to the desired COL. With attractive physico-chemical properties of SiC and large specific surface area of nanoporous SiC–C composites, we expected that there would be special interaction between Pt nanoparticles and nanoporous SiC–C composites and so that the surface electronic state of Pt nanoparticles might be modulated and thus be beneficial for preferential adsorption and activation of CAL to form the desired product COL.
O double bond energy than that of CC double bond.21 With regards of the selective hydrogenation of CAL, special supports such as graphene or carbon nanotubes for loading monometallic Pt catalyst21–27 or bimetallic catalysts (such as Pt–Fe)21,28,29 are usually required to achieve high selectivity to the desired COL. With attractive physico-chemical properties of SiC and large specific surface area of nanoporous SiC–C composites, we expected that there would be special interaction between Pt nanoparticles and nanoporous SiC–C composites and so that the surface electronic state of Pt nanoparticles might be modulated and thus be beneficial for preferential adsorption and activation of CAL to form the desired product COL.
 | ||
| Scheme 1 Selective hydrogenation of CAL. | ||
Experimental
Catalyst preparation
The monometallic 5 wt% Pt/SiC–C catalyst was prepared using an ultrasound-promoted impregnation method. In a typical process, 0.4218 g SiC–C powder was firstly treated under vacuum for 10 minutes at 90 °C. Then, SiC–C was rapidly added to 2 mL aqueous solution of chloroplatinic acid containing 22.2 mg Pt followed by a vigorous stirring for about 1 min to ensure that the solid powder was mixed well with liquid quickly. The SiC–C was impregnated with the Pt precursor in an ultrasonic instrument for 4 h. Then, the catalyst precursor was drying at 80 °C to remove the excess solvent. Finally, the catalyst precursor was reduced in an aqueous solution of sodium formate at a refluxed temperature (95 °C) and washed with plenty of water to remove chlorine ions.For comparison, monometallic 5 wt% Pt catalysts supported on other materials, such as SBA-15 silica, CMK-3 carbon replicated from SBA-15, and S16-C carbon replicated from SBA-16 silica, were also prepared using the same method as the Pt/SiC–C catalyst. SBA-15 silica, CMK-3 carbon and SBA-16 silica were prepared according to ref. 30–32. S16-C carbon was replicated using SBA-16 silica as a hard template and sucrose as carbon source according to the method with which CMK-3 carbon was prepared. In addition, 5 wt% Pt/C and Pt/Al2O3 catalysts were also purchased from Alfa Aesar as reference catalysts.
Catalyst characterization
The catalyst was characterized in detail with many physico-chemical techniques. The XRD patterns of samples were collected on a Bruker D8 Advance instrument using Cu-Kα radiation. The nitrogen adsorption–desorption isotherms were measured at −196 °C (77 K) on a Quantachrome Autosorb-3B system, after the samples were evacuated for 10 h at 200 °C. The BET specific surface area was calculated using adsorption data in the relative pressure range from 0.05 to 0.30. The pore size distributions were calculated from the analysis of the adsorption branch of the isotherm using the BJH algorithm. The SEM images were taken on a Hitachi S4800 electro-microscope with an acceleration voltage of 20 kV. The TEM images were taken on an FEI Tecnai G2-TF30 microscope at an acceleration voltage of 300 kV. The Pt loading on samples and the leached Pt amount in filtrate was detected with a Thermo Elemental IRIS Intrepid II XSP inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The thermogravimetric (TG) analysis of the samples was conducted from r.t. to 800 °C under air atmosphere with Mettler Toledo TGA/SDTA851e apparatus. The IR spectrum was measured with a Nicolet NEXUS 670 spectrometer. The Raman spectrum of sample was taken on a Renishaw inVia Raman Spectrometer excited using a 514 nm laser.To explain the superiority of SiC–C supported Pt catalyst towards the selective hydrogenation of CAL, the Pt/SiC–C catalyst was further characterized using XPS with a Thermo Fisher Scientific ESCALAB 250Xi spectrometer with Al Kα radiation (1486.6 eV) as incident beam with a monochromator. The sample was in situ pre-treated in flowing hydrogen at 400 °C for 1 h in a reactor attachment of the XPS spectrometer. The binding energy (BE) was calibrated using C–C binding energy at 284.4 eV in order to compare the binding energies with the data from the literatures. The spectra shown in the figures have been corrected by subtraction of a Shirley background. Spectral fitting and peak integration was done using the XPSPEAK software.
Catalytic reaction
In a typical reaction, 0.03 g 5 wt% Pt/support was pre-treated under a flowing hydrogen (40 mL min−1) at 400 °C for 2 h before use. The catalyst was then transferred to a 100 mL autoclave and mixed with solvent (18 mL isopropanol and 2 mL water) and substrate (960 μL CAL). The reaction began when hydrogen (2.0 MPa) was introduced with stirring (1200 rpm) at a required temperature. The reaction was stopped after 1 h and the products were analyzed by GC-FID (GC-2014, Shimadzu) equipped with a capillary column (DM-WAX, 30 m × 0.25 mm × 0.25 μm). The response factor of each component was calculated using standard samples and was used to calculate the conversion and selectivity. After each run, the catalyst was recovered by centrifugation and washed with fresh solvent for several times. Then, fresh reactant and solvent were charged to the autoclave together with the recovered catalyst to conduct the next run reaction.Results and discussion
Catalyst characterization
The nanoporous SiC–C composites were purchased from Suzhou Yuhao Nanomaterials Inc. (Suzhou, China). The composites were detected by TG analysis under an air atmosphere to determine the composition and the weight ratio of C/SiC was about 1.2 (Fig. S1, ESI†). The 5 wt% Pt/SiC–C catalyst was prepared by an ultrasound promoted impregnation method followed by reduction in an aqueous solution of sodium formate. According to the ICP-AES result, the final Pt loading in the Pt/SiC–C catalyst was the same as the nominal value. This demonstrates that the interaction between Pt nanoparticles and SiC–C composites was extremely high so that the Pt nanoparticles could not be washed off from the support surface during the liquid-phase reduction at a refluxed temperature (95 °C). The liquid-phase selective hydrogenation of CAL was carried out in a stainless-steel autoclave equipped with a heater and a magnetic stirrer.The structure and crystalline phase of nanoporous SiC–C composites and Pt/SiC–C catalyst was firstly characterized using XRD. As displayed in Fig. 1(a), nanoporous SiC–C composites gave typical diffraction patterns associated to (111), (220) and (311) planes of β-SiC besides weak stacking faults at 2 theta angle of 35°.11 In addition, the diffraction peak assigned to amorphous carbon was also observed at around 2 theta of 25°. The β-SiC crystalline phase was well retained in the final Pt/SiC–C catalyst, which was confirmed by the XRD pattern of Pt/SiC–C catalyst. Additionally, the Pt/SiC–C catalyst also gave typical but weak diffraction peaks assigned to Pt(111), Pt(200) and Pt(220) planes, indicating that the Pt particles were well dispersed on the nanoporous SiC–C composites surface without formation of large aggregation.
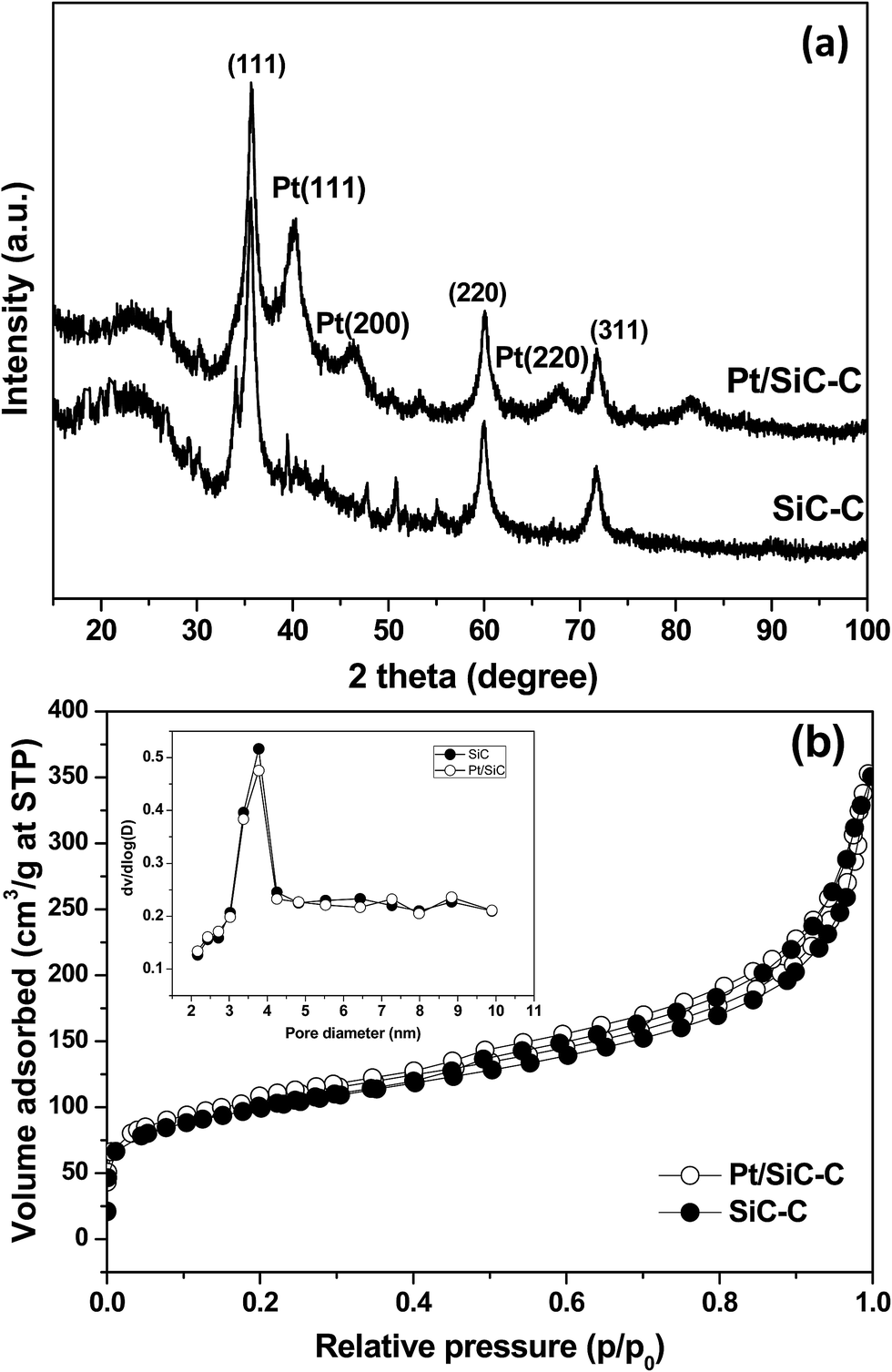 | ||
| Fig. 1 (a) XRD patterns and (b) nitrogen adsorption–desorption isotherms of nanoporous SiC–C and Pt/SiC–C catalyst. | ||
The porous structure and specific surface area of the SiC–C composites and the Pt/SiC–C catalyst was also determined using nitrogen sorption (Fig. 1(b)). Both the SiC–C composites and the Pt/SiC–C catalyst showed type IV isotherms, suggesting there are mesoporous pores although the hysteresis loops were not so large. The pore size of Pt/SiC–C and SiC–C centered at 3.6 nm (inset of Fig. 1(b)). For clarity, Table 1 lists the detailed physicochemical parameters of the SiC–C composites and the Pt/SiC–C catalyst. The BET specific surface area of Pt/SiC–C was comparable with that of SiC–C composites, having a large specific surface area of more than 330 m2 g−1. The pore volume of the Pt/SiC–C catalyst was also comparable to that of SiC–C composites, demonstrating the deposition of Pt nanoparticles did not block the pore entrance of SiC–C composites. We also characterized the Pt/SiC–C catalyst using Raman and infrared spectroscopy. As displayed in Fig. S2,† the Pt/SiC–C catalyst showed a typical Si–C vibration band at around 835 cm−1, while for the Raman spectra of SiC–C composites and Pt/SiC–C catalyst, only G and D bands assigned to sp2 hybridized carbon were observed and the typical Si–C vibration band was not detected (Fig. S3†). It indicates that SiC was covered by carbon with thick layers, so that the signal of SiC could not be detected.
| Sample | SBET (m2 g−1) | Vp (cm3 g−1) | d (nm) |
|---|---|---|---|
| SiC–C | 334 | 0.51 | 3.6 |
| Pt/SiC–C | 346 | 0.52 | 3.6 |
The morphology of Pt/SiC–C catalyst was characterized using SEM (Fig. 2(a) and (b)). The SiC–C composites exhibited irregular shapes with broccoli-like aggregation. The Pt particle size distribution of the Pt/SiC–C catalyst was also characterized using TEM. As exhibited in Fig. 2(c), Pt nanoparticles were uniformly dispersed on the SiC–C surface for the fresh Pt/SiC–C catalyst. The Pt particle size distribution for the fresh Pt/SiC–C catalyst was also displayed in Fig. 2(d). Accordingly, the Pt particle size for the fresh Pt/SiC–C catalyst varied in the range of 1.4–2.8 nm and the average Pt particle size was 2.1 nm with 52.3% dispersion.
 | ||
| Fig. 2 (a and b) SEM images of Pt/SiC–C catalyst, (c and d) TEM image and Pt particle size distribution of fresh Pt/SiC–C catalyst, (e and f) TEM image and Pt particle size distribution of the used Pt/SiC–C catalyst after 10 cycles. | ||
Catalytic performance of the Pt/SiC–C catalyst
With high and uniform dispersion of Pt nanoparticles on SiC–C composites, the Pt/SiC–C catalyst was anticipated to work well in the liquid-phase hydrogenation of CAL. We firstly investigated the kinetic profiles of selective hydrogenation of CAL with the Pt/SiC–C catalyst at different temperatures. As revealed in Fig. 3(a), when the reaction was conducted at 25 °C, a 32.8% conversion was obtained within an initial 15 min. The conversion of CAL was gradually increased with reaction time and reached 84.9% after 1 h as a result. Based on the conversion obtained within the initial 15 min, the activity (represented by TOF, defined as the number of moles of converted CAL per mole of Pt active sites per hour) could reach 2446 h−1. It is worth noting that COL was the primary product with about 80% selectivity during the whole process. In order to evaluate temperature effect on the selective hydrogenation of CAL, the reaction was also investigated with the Pt/SiC–C catalyst at 40 °C. Undoubtedly, the reaction went slightly faster at 40 °C when compared with that conducted at 25 °C. A 50.2% conversion was afforded within an initial 15 min, and almost complete conversion of CAL was achieved after 1 h at 40 °C. Although the conversion of CAL was obviously increased with the temperature and the TOF could reach 3745 h−1 at 40 °C, it seemed that the selectivity to COL was independent of the reaction temperature. That is, even the temperature was increased to 40 °C, the selectivity to COL was not decreased with temperatures and as a result, about 80% selectivity to COL was still achieved by the Pt/SiC–C catalyst. | ||
| Fig. 3 (a) Kinetic profiles of liquid-phase selective hydrogenation of CAL with Pt/SiC–C catalyst at 25 °C and 40 °C, respectively. Reaction conditions: 0.03 g Pt catalyst, 20 mL solvent (18 mL isopropanol and 2 mL water), 960 μL CAL, 2.0 MPa H2, 1200 rpm. (b) Temperature effect on liquid-phase hydrogenation of CAL. Reaction conditions: 0.03 g Pt catalyst, 20 mL solvent (18 mL isopropanol and 2 mL water), 960 μL CAL, 2.0 MPa H2, 1200 rpm, 1 h. | ||
In order to further investigate the temperature effect on selectivity to COL, the reaction was conducted at elevated temperatures. As clearly displayed in Fig. 3(b), although the conversion was further increased with the temperature, the selectivity to COL was still kept about 80%. That is, the reaction temperature only influenced the reaction activity and the conversion of CAL was almost linearly increased with temperature, while the selectivity to COL was independent of the temperature. Even at 80 °C, the selectivity to COL was also around 80%. Nevertheless, taking operating cost into account, in the following studies, room temperature (25 °C) was still adopted as the standard reaction conditions. In addition, according to the results obtained with the Pt/SiC–C catalyst at different temperatures, the Pt/SiC–C catalyst was very active and highly selective for the liquid-phase hydrogenation of CAL, even at room temperature.
On the contrary, when we made a literature survey about the liquid-phase selective hydrogenation of CAL, we noticed that the similar reaction was usually performed at relatively higher temperatures (40–95 °C).22–29,33 Even those reactions were performed at higher temperatures, most results including activity and selectivity to COL obtained in literatures were lower than those we obtained with the Pt/SiC–C catalyst even at room temperature in this study. Moreover, many researches demonstrated that the monometallic Pt catalysts were seldom selective for the hydrogenation of CO bond due to thermodynamic reason.21 In order to compare the catalytic performance of monometallic Pt catalysts supported on different materials for the selective hydrogenation of CAL under our tested conditions, 5 wt% Pt catalysts supported on other materials, such as ordered mesoporous silica SBA-15, ordered mesoporous carbon CMK-3 replicated from SBA-15 silica, and ordered mesoporous carbon S16-C replicated from SBA-16 silica, were prepared using the similar method and investigated for the tested reaction. The commercial 5 wt% Pt/C and Pt/Al2O3 catalysts purchased from Alfa Aesar were also applied as reference catalysts.
As shown in Fig. 4(a), the conversion and selectivity to the desired product COL varied with different materials for loading Pt nanoparticles. The conversion of CAL increased with the order: Pt/SBA-15 < Pt/S16-C ≈ Pt/C ≈ Pt/Al2O3 < Pt/CMK-3 < Pt/SiC–C. Among the Pt catalysts tested for this study, the nanoporous SiC–C composites supported Pt catalyst was most active under the same conditions. As for the selectivity to COL afforded by different Pt catalysts, the sequence follows the order as: Pt/Al2O3 < Pt/CMK-3 < Pt/SBA-15 ≈ Pt/S16-C < Pt/C < Pt/SiC–C. The Pt/SBA-15 and Pt/S16-C catalysts gave comparable selectivity to COL and HCAL (hydrocinnamaldehyde), suggesting that the Pt/SBA-15 and Pt/S16-C were not selective either for CO bond or for CC bond hydrogenation. While for the Pt/CMK-3 and Pt/Al2O3 catalysts, they were selective for CC bond hydrogenation and the main product HCAL was afforded. Although simultaneous hydrogenation of both CO and CC double bond was inevitable because HCOL (hydrocinnamyl alcohol) was also detected on each Pt catalyst supported on whatever material, the content of HCOL was low. For clarity, Fig. 4(b) shows the product distribution obtained with different Pt catalysts under the same conditions. As can be clearly seen, the Pt/SiC–C catalyst gave the prominent product COL, while CAL, HCAL and HCOL are minor components. For other Pt catalysts supported on silica, carbon or Al2O3, there was no priority in product distribution.
 | ||
| Fig. 4 (a) Conversions and selectivity obtained with Pt catalysts supported on different materials and (b) the products distribution obtained on monometallic Pt catalysts supported on different materials toward the liquid-phase hydrogenation of CAL at room temperature. 5 wt% Pt/C and Pt/Al2O3 were purchased from Alfa Aesar and other 5 wt% Pt catalysts were prepared using a same method as the Pt/SiC–C catalyst. Reaction conditions: 0.03 g Pt catalyst, 20 mL solvent (18 mL isopropanol and 2 mL water), 960 μL CAL, 2.0 MPa H2, 25 °C, 1200 rpm, 1 h. | ||
Further characterization of the Pt/SiC–C catalyst using XPS
It suggests that monometallic Pt catalysts supported on traditional supports such as carbon, silica and alumina are indeed unselective for the liquid-phase hydrogenation of CAL; while the monometallic Pt catalyst supported on SiC–C composites is not only active, but also highly selective for production of COL during the liquid-phase hydrogenation of CAL. To explain the superiority of nanoporous SiC–C composites supported Pt catalyst towards the selective hydrogenation of CAL, the Pt/SiC–C catalyst was further characterized using XPS after in situ pre-treated in flowing hydrogen at 400 °C for 1 h in a reactor attachment of the XPS spectrometer. For C1s spectrum of Pt/SiC–C catalyst, the binding energy (BE) at 283.1 eV could be ascribed to C–Si of SiC, while the BE at 284.4 eV undoubtedly belongs to C–C of amorphous carbon. In addition, there was trace amount of oxygen-containing groups on SiC–C composite surface (Fig. 5(a)). As for Si species on SiC–C composite surface, Si–C species are dominant on the surface, in accompany with a little silica (Fig. 5(b)). The Pt4f spectrum could be deconvoluted to three Pt species (Fig. 5(c)). The BE of 71.2 eV could be assigned to 4f7/2 of Pt0 species, while 72.0 eV and 74.0 eV were ascribed to 4f7/2 of Pt2+ and Pt4+ species, respectively.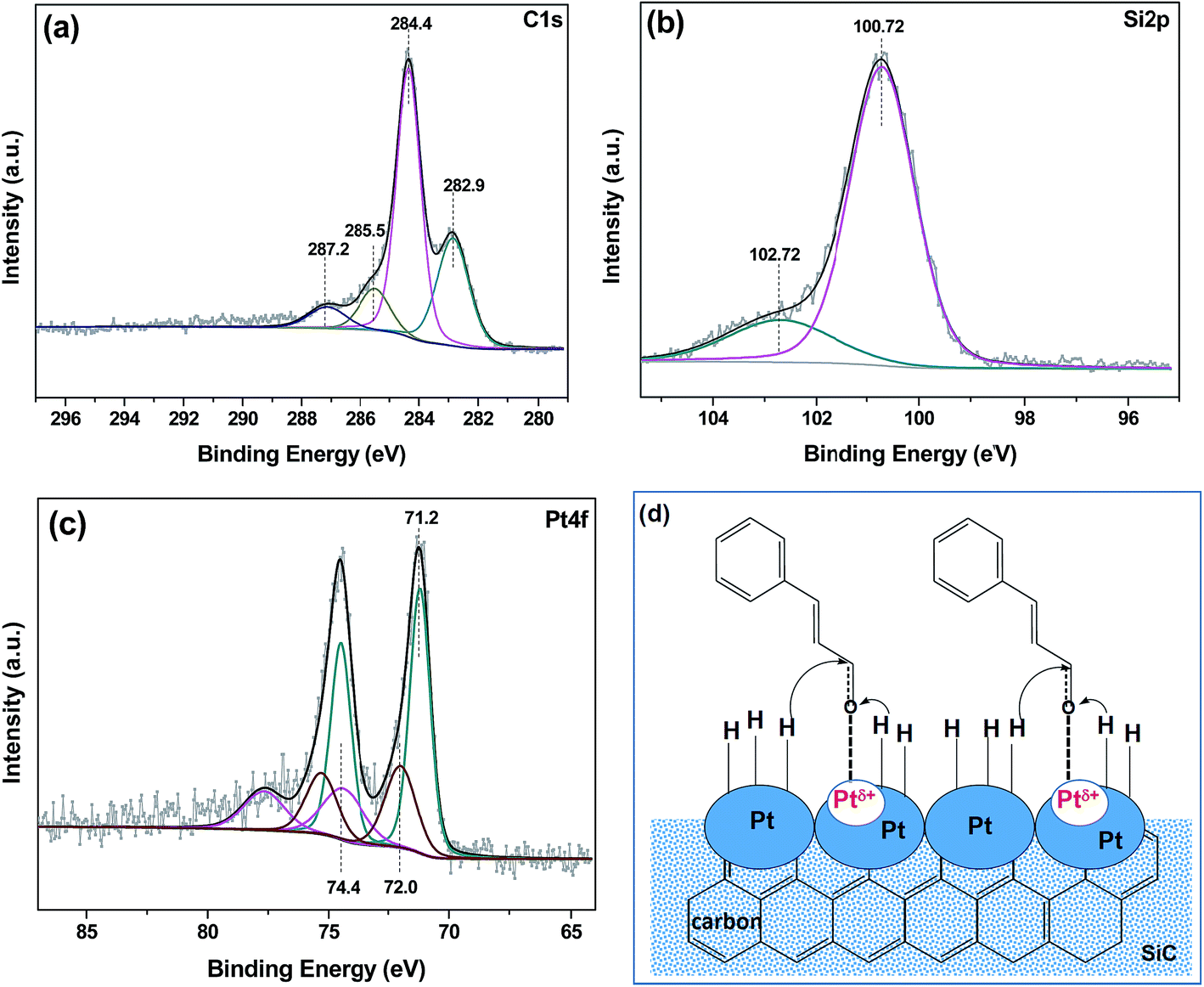 | ||
| Fig. 5 (a) C1s XPS, (b) Si2p XPS, (c) Pt4f XPS spectra of Pt/SiC–C catalyst and (d) the hypothesis for CAL adsorption and activation with the Pt/SiC–C catalyst. | ||
Based on the Pt4f characterization results, although Pt0 species were dominant, there were still quite a few Pt species with high valences Ptδ+ (Pt2+ or Pt4+) on the Pt/SiC–C surface. It should be emphasized that the sample had been in situ pre-treated under a flowing hydrogen atmosphere after reduction in an aqueous solution of sodium formate. This verifies that there is strong interaction between Pt species and the SiC–C composites and the electron transfer occurs from Pt species to SiC–C composites. We deduce that the Pt species with positive charge would be beneficial for preferential adsorption of carbonyl bond of CAL via electrostatic interaction between Ptδ+ species and carbonyl oxygen atom, so that the activation of carbonyl bond was improved. Meanwhile, hydrogen atoms via dissociative adsorption on Pt0 species can attack the activated carbonyl group of CAL to form the desired product COL. As a result, high selectivity to COL was afforded with the Pt/SiC–C catalyst. The hypothesis for adsorption, activation and hydrogenation of CO double bond of CAL was also proposed in Fig. 5(d).
Reusability of the Pt/SiC–C catalyst
The reusability is one of the important matters to be considered for a heterogeneous catalyst involved in a liquid-phase reaction. Therefore, we investigated the reusability of the Pt/SiC–C catalyst toward the liquid-phase hydrogenation of CAL at room temperature. After the reaction, the Pt/SiC–C catalyst was filtered and washed using fresh solvent and then participated in the next run. To our delight, the Pt/SiC–C catalyst can be easily recovered and recycled for at least 10 runs without any loss in activity or selectivity to COL (Fig. 6). Moreover, we noticed that even higher CAL conversions and COL selectivity were furnished with the used Pt/SiC–C catalyst after the first run. Meanwhile, the selectivity to HCAL was suppressed as well (see Fig. S4 for the detailed product distribution†). It can be interpreted by that the catalyst surface was reconstructed after the first run and the catalyst surface was more suitable for the selective hydrogenation of CAL to the desired product COL. We also detected the filtrate using ICP-AES to ensure the heterogeneous reaction. As a result, the leached Pt amount was below the detection limit of ICP-AES. This further demonstrates that Pt nanoparticles supported on SiC–C surface are stable enough during the recycling processes. In addition, we also characterized the used Pt/SiC–C catalyst using TEM. As already displayed in Fig. 2(e), the Pt nanoparticles were still uniformly dispersed on the SiC–C surface except that the Pt particles were slightly aggregated after 10 reaction cycles. According to the Pt particle size distribution in Fig. 2(f), the Pt particle size of the used Pt/SiC catalyst was centered in 2.2–2.6 nm, with an average Pt particle size of 2.4 nm.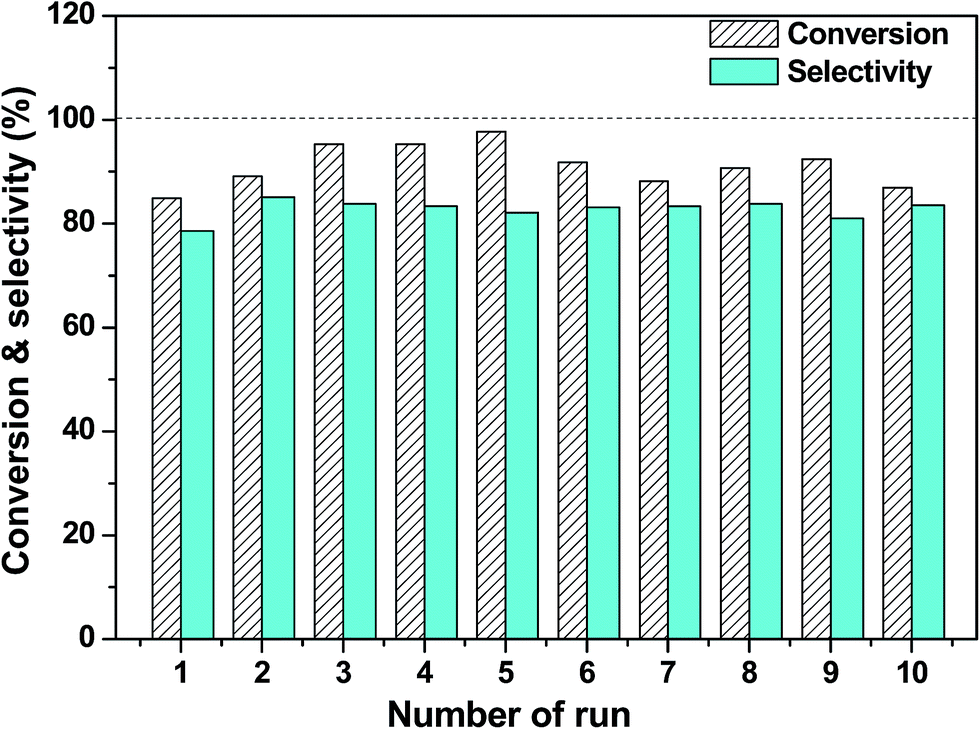 | ||
| Fig. 6 The reusability of the Pt/SiC–C catalyst toward the liquid-phase hydrogenation of CAL. Reaction conditions: 0.03 g Pt/SiC–C catalyst, 20 mL solvent (18 mL isopropanol and 2 mL water), 960 μL CAL, 2.0 MPa H2, 25 °C, 1200 rpm, 1 h. | ||
Conclusions
In summary, the Pt catalyst supported on nanoporous SiC–C composites was developed for the liquid-phase selective hydrogenation of CAL. To our delight, the monometallic Pt catalysts supported on nanoporous SiC–C composites was proved to be active and highly selective for the liquid-phase hydrogenation of CAL. Furthermore, the Pt/SiC–C catalyst showed superior catalytic performance to Pt catalysts supported on other materials such as silica, carbon and alumina. Of particular note is that the Pt/SiC–C catalyst could be easily recycled for at least 10 runs without any loss in activity or selectivity to the desired product. The leached Pt amount in the filtrate was below the detection limit of ICP-AES. The Pt nanoparticles were still well dispersed on the support surface even after 10 reaction runs. It is deduced that the Pt particles with positive charges, derived from strong interaction between Pt particles and SiC–C composites, are beneficial for preferential adsorption and activation of carbonyl bond of CAL via electrostatic interaction between oxygen atoms of carbonyl group and Pt species with positive charges.Acknowledgements
The research was financially supported by the National Natural Science Foundation of China (Grant Nos. 21273076 and 21373089), the Open Research Fund of Top Key Discipline of Chemistry in Zhejiang Provincial Colleges and Key Laboratory of the Ministry of Education for Catalysis Materials (Zhejiang Normal University) (ZJHX2013), and Shanghai Leading Project (B409).References
- H. Morkoç, S. Strite, G. B. Gao, M. E. Lin, B. Sverdlov and M. Burns, J. Appl. Phys., 1994, 76, 1363 CrossRef.
- R. Presser and K. G. Nickel, Crit. Rev. Solid State Mater. Sci., 2008, 33, 1 CrossRef.
- R. Wu, K. Zhou, C. Y. Yue, J. Wei and Y. Pan, Prog. Mater. Sci., 2015, 72, 1 CrossRef CAS.
- H. Liu, G. She, L. Mu and W. Shi, Mater. Res. Bull., 2012, 47, 917 CrossRef CAS.
- J. Hao, Y. Y. Wang, X. L. Tong, G. Q. Jin and X. Y. Guo, Int. J. Hydrogen Energy, 2012, 37, 15038 CrossRef CAS.
- J. J. Yang, X. P. Zeng, L. J. Chen and W. X. Yuan, Appl. Phys. Lett., 2013, 102, 083101 CrossRef.
- X. Shen and S. T. Pantelides, J. Phys. Chem. Lett., 2013, 4, 100 CrossRef CAS PubMed.
- X. F. Liu, M. Antonietti and C. Giordano, Chem. Mater., 2013, 25, 2021 CrossRef CAS.
- L. Dong, J. Zang, J. Su, Y. Jian, Y. Wang, J. Lu and X. Xu, Int. J. Hydrogen Energy, 2014, 39, 16310 CrossRef CAS.
- J. J. Niu and J. N. Wang, Acta Mater., 2009, 57, 3084 CrossRef CAS.
- L. Fang, X. P. Huang, F. Vidal-Iglesias, Y. P. Liu and X. L. Wang, Electrochem. Commun., 2011, 13, 1309 CrossRef CAS.
- H. Dai, Y. Chen, Y. Lin, G. Xu, C. Yang, Y. Tong, L. Guo and G. Chen, Electrochim. Acta, 2012, 85, 644 CrossRef CAS.
- R. Dhiman, E. Johnson, E. M. Skou, P. Morgen and S. M. Andersen, J. Mater. Chem. A, 2013, 1, 6030 CAS.
- Y. Liu, I. Florea, O. Ersen, C. Pham-Huu and C. Meny, Chem. Commun., 2015, 51, 145 RSC.
- Y. Liu, O. Ersen, C. Meny, F. Luck and C. Pham-Huu, ChemSusChem, 2014, 7, 1218 CrossRef CAS PubMed.
- H. Ba, Y. Liu, X. Mu, W.-H. Doh, J.-M. Nhut, P. Granger and C. Pham-Huu, Appl. Catal., A, 2015, 499, 217 CrossRef CAS.
- H. Liu, J. Diao, Q. Wang, S. Su, T. Chen, C. Miao, W. Yang and D. Su, Chem. Commun., 2014, 50, 7810 RSC.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014, 5, 3688 CAS.
- Y. Cui, X. Guo, Y. Wang and X. Guo, Chin. J. Catal., 2015, 36, 322 CrossRef CAS.
- L. Truong-Phuoc, T. Truong-Huu, L. Nguyen-Dinh, W. Baaziz, T. Romero, D. Edouard, D. Begin, I. Janowska and C. Pham-Huu, Appl. Catal., A, 2014, 469, 81 CrossRef CAS.
- Y. Yuan, S. Yao, M. Wang, S. Lou and N. Yan, Curr. Org. Chem., 2013, 17, 400 CrossRef CAS.
- Z. Tian, Q. Li, J. Hou, L. Pei, Y. Li and S. Ai, J. Catal., 2015, 331, 193 CrossRef CAS.
- S. Handjani, E. Marceau, J. Blanchard, J.-M. Krafft, M. Che, P. Maki-Arvela, N. Kumar, J. Warna and D. Yu. Murzin, J. Catal., 2011, 282, 228 CrossRef CAS.
- X. Ji, X. Niu, Q. Han, F. Yuan, F. Zaera, Y. Zhu and H. Fu, ChemCatChem, 2014, 6, 3426 CrossRef.
- Q. Wu, C. Zhao, B. Zhang, X. Li, Z. Ying, T. Liu, W. Lin, Y. Yu, H. Cheng and F. Zhao, J. Colloid Interface Sci., 2016, 463, 75 CrossRef CAS PubMed.
- J. Shi, R. Nie, P. Chen and Z. Hou, Catal. Commun., 2013, 41, 101 CrossRef CAS.
- Z. Sun, Z. Rong, Y. Wang, Y. Xia, W. Du and Y. Wang, RSC Adv., 2014, 4, 1874 RSC.
- K. B. Vu, K. V. Bukhryakov, D. H. Anjum and V. O. Rodionov, ACS Catal., 2015, 5, 2529 CrossRef CAS.
- G. Neri, I. Arrigo, F. Corigliano, L. De Luca and A. Donato, Catal. Lett., 2011, 141, 1590 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
- S. Jun, S. Joo, R. Ryoo and M. Kruk, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- T.-W. Kim, R. Ryoo, M. Kruk, K. P. Gierszal, M. Jaroniec, S. Kamiya and O. Terasaki, J. Phys. Chem. B, 2004, 108, 11480 CrossRef CAS.
- B. F. Machado, S. Morales-Torres, A. F. Perez-Cadenas, F. J. Maldonado-Hodar, F. Carrasco-Marin, A. M. T. Silva, J. L. Figueiredo and J. L. Faria, Appl. Catal., A, 2012, 425–426, 161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15164g |
| This journal is © The Royal Society of Chemistry 2016 |