
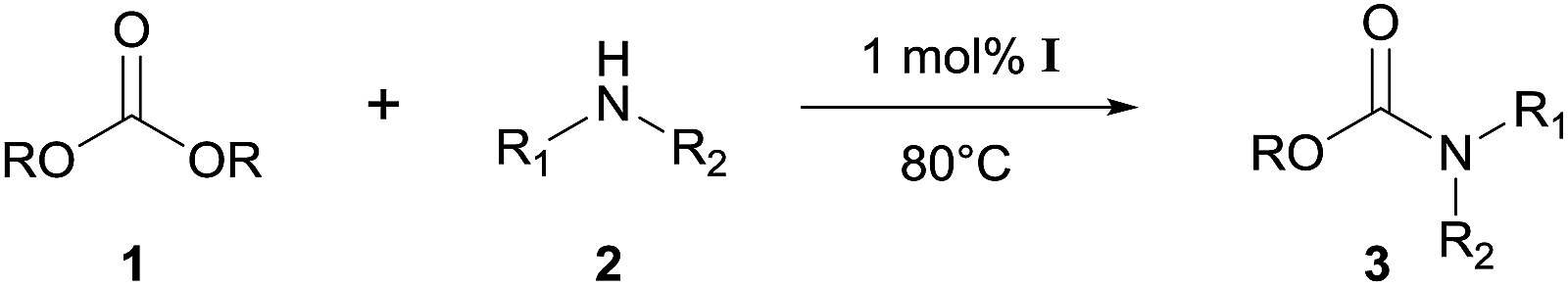
Heterobimetallic dinuclear lanthanide alkoxide complexes as acid–base bifunctional catalysts for synthesis of carbamates under solvent-free conditions†
Ruijie Zeng‡
,
Linquan Bao‡,
Hongting Sheng*,
Lili Sun,
Man Chen,
Yan Feng* and
Manzhou Zhu
School of Chemistry and Chemical Engineering, Center for Atomic Engineering of Advanced Materials, Anhui University, Hefei 230039, P. R. China. E-mail: sht_anda@126.com; fy70@163.com; Fax: +86 551 63861487; Tel: +86 551 63861487
First published on 9th August 2016
Abstract
Heterobimetallic dinuclear lanthanide alkoxide complexes Ln2Na8(OCH2CH2NMe2)12(OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)] were used as efficient acid–base bifunctional catalysts for the synthesis of carbamates from dialkyl carbonates and amines as well as the N-Boc protection of amines. The cooperative catalysts showed high catalytic activity and a wide scope of substrates with good to excellent yields under solvent-free conditions. The systems have shown higher catalytic activities due to the noteworthy synergistic interactions of Lewis acid center–Brønsted basic center. The comparison of catalytic efficiency between mono- and dinuclear heterobimetallic lanthanide alkoxide analogues was also investigated.
Introduction
Organic carbamates are crucial intermediates in the synthesis of pharmaceuticals, pesticides, herbicides, fungicides and commodity chemicals.1 Previously, carbamates were prepared from the action of corresponding amines with phosgene or its derivatives.2 These reactions have resulted in serious environment and safety problems due to the extreme toxicity of phosgene and HCl produced as the main by-product. To overcome this drawback, great efforts have been made to synthesize carbamates using other reagents for the replacement of phosgene, such as oxidative carbonylation of aromatic amines,3 catalytic reductive carbonylation of nitroaromatics,4 coupling reactions of amines and carbon dioxide with a different third component (such as alcohols, alkyl halides, tosylhydrazones or diaryliodonium salts),5 treatment of alcohols and thiols with cyanate salts,6 reacting azidoformates with boronic acids,7 rearrangement reactions,8 and C–H bond functionalization9 etc.Compared with the conventional methods, synthesis of carbamates from amines and dialkyl carbonates is considered as one of the most promising methods for its environmental friendly and nontoxic nature, and potential industrial applications.10 Since the only by-product of this reaction is alcohol, which could be removed easily from the reaction, the usage of dialkyl carbonates is an alternative to phosgene or chloroformate. Recently, several methods for the synthesis of carbamates from dialkyl carbonates and amines have been reported.10,11 However, these methodologies may present some disadvantages such as high cost,11d high temperature10c,10d,11c,11e or pressure,10d,11c high toxicity,10a,11f large excess amounts of dialkyl carbonates and narrowed scope of amines.10c,11c–11e
Lanthanide complexes have shown better catalytic properties in many organic reactions.12 Heterobimetallic lanthanide complex as an important family of lanthanide complexes has been developed over the last two decades.13 Shibasaki and co-workers developed ground breaking lanthanide-alkali metal heterobimetallic complexes M3[Ln(BINOL)3] (where Ln = rare earth metal, M = Li, Na, K, and BINOL is 1,1′-bis(2-naphthol)), which exhibited both Lewis acidity/Brønsted basicity bifunctional catalysis in a variety of stereoselective organic transformations due to the cooperativity of catalysts.14 Recent advances from their research on cooperative asymmetric catalysis were focused on dinuclear Schiff base catalysis.15 For instance, heterobimetallic Cu/Sm–Schiff base complex catalyze daza-Henry reaction with high syn-selectivity.16 Besides, the highly efficient heterogeneous Nd/Na heterobimetallic catalyst was successfully applied to asymmetric nitroaldol reaction.17
As a part of our ongoing studies on heterometallic rare earth-alkali metal catalysts,18 we previously found that heterobimetallic rare earth-alkali metal alkoxide complexes were the efficient catalysts for ring-opening polymerization of ε-caprolactone and trimethylene carbonate.18a,18c,18d Recently, we have reported that heterobimetallic dinuclear lanthanide alkoxide complexes as acid–base bifunctional catalysts were successfully applied to the transesterification under mild conditions.18g In this system, the dinuclear lanthanide catalysts acted as not only binary Lewis acid centers to activate esters, but also multi-Brønsted base centers to activate alcohols, which was important to achieve high activity (Fig. 1). Currently, Kazushi and Takashi group reported the transesterification of β-keto esters by tetranuclear zinc cluster catalyst. High catalyst activity depended on the balance between lewis acidity and Brønsted basicity, it is consist with the dual activation by the cooperative zinc centers.19
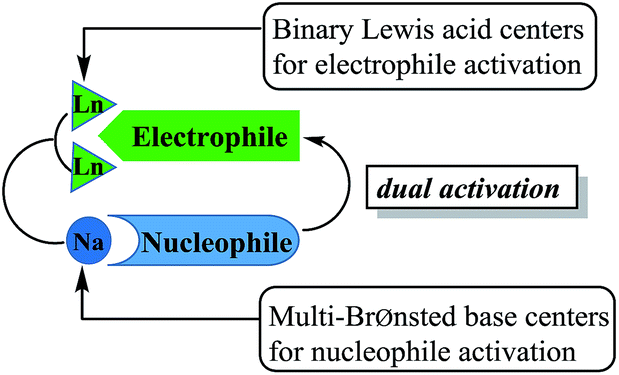 | ||
| Fig. 1 Dual activation by the heterobimetallic dinuclear lanthanide complex as an acid–base bifunctional catalyst. | ||
On the basis of the facts mentioned above, we attempted to perform the synthesis of carbamates through the dual activation of carbonates and amines by the heterobimetallic dinuclear lanthanide alkoxide complexes. So, we report an efficient method for the synthesis of carbamates from dialkyl carbonates and amines, as well as the N-Boc protection of amines under solvent-free conditions in this work. Meanwhile, the comparison of catalytic efficiency between readily available heterobimetallic mononuclear lanthanide alkoxide analogues LnNa8(OtBu)10(OH) [Ln: i (Nd) and ii (Yb)] and above dinuclear catalysts Ln2Na8(OCH2CH2NMe2)12(OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)] (Fig. 2), was also investigated.
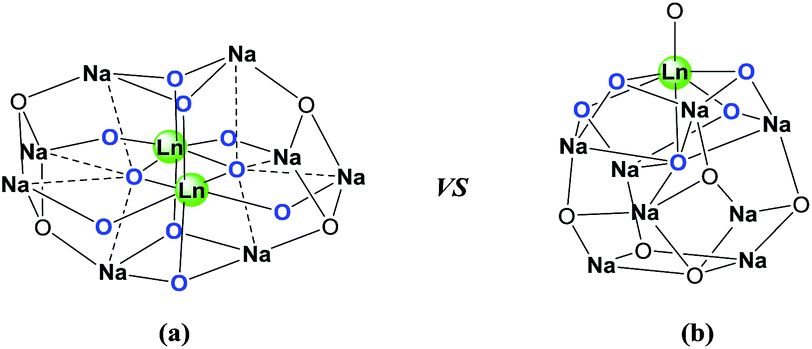 | ||
| Fig. 2 (a) Structural framework of heterobimetallic dinuclear lanthanide alkoxide complexes Ln2Na8(OCH2CH2NMe2)12(OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)].18a (b) Structural framework of heterobimetallic mononuclear lanthanide alkoxide analogues LnNa8(OtBu)10(OH) [Ln: i (Nd) and ii (Yb)].18d | ||
Results and discussion
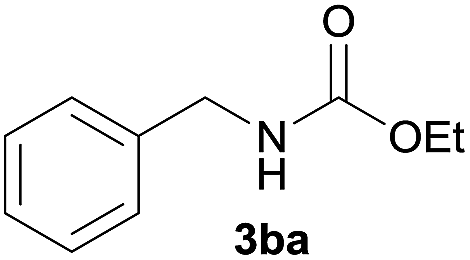
The synthesis of carbamate 3aa from dimethyl carbonate 1a (DMC) and benzyl amine 2a was chosen as a model system (Table 1). The optimal reaction conditions, including various catalysts, different catalyst loadings, and diverse solvents for the reaction process were investigated in detail. Four kinds of alkoxide complexes, including heterobimetallic dinuclear lanthanide alkoxide complexes Ln2Na8(OCH2CH2NMe2)12 (OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)], monometallic lanthanide alkoxide complex Nd(OCH2CH2NMe2)3, monometallic sodium alkoxide complex NaOCH2CH2NMe2 and heterobimetallic mononuclear lanthanide alkoxide analogues LnNa8(OtBu)10(OH) [Ln: i (Nd) and ii (Yb)] were chosen to study the difference of catalytic activity between monometallic vs. heterobimetallic, and heterobimetallic mono- vs. dinuclear lanthanide catalysts. The above alkoxide complexes were synthesized according to the literature methods.18 And X-ray crystal structures of Ln2Na8(OCH2CH2NMe2)12(OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)] and LnNa8(OtBu)10(OH) [Ln: i (Nd) and ii (Yb)] were also reported,18a,d whose structural frameworks are shown in Fig. 2.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Loading of catalyst (mol%) | Solvent | Yieldb (%) |
| a Reaction conditions: dimethyl carbonate 1a (2 mmol), benzyl amine 2a (2 mmol) and catalyst at 80 °C under an argon atmosphere for 12 h.b GC yield.c OR = OCH2CH2NMe2. | ||||
| 1 | — | — | Toluene | 8 |
| 2 | I | 1.0 | Toluene | 85 |
| 3 | II | 1.0 | Toluene | 77 |
| 4 | III | 1.0 | Toluene | 71 |
| 5 | IV | 1.0 | Toluene | 72 |
| 6 | Nd(OR)3c | 2.0 | Toluene | 18 |
| 7 | NaORc | 8.0 | Toluene | 49 |
| 8 | i | 1.0 | Toluene | 66 |
| 9 | ii | 1.0 | Toluene | 54 |
| 10 | I | 0.5 | Toluene | 76 |
| 11 | I | 1.5 | Toluene | 87 |
| 12 | I | 2.0 | Toluene | 90 |
| 13 | I | 1.0 | DMF | 55 |
| 14 | I | 1.0 | DMSO | 60 |
| 15 | I | 1.0 | Acetonitrile | 61 |
| 16 | I | 1.0 | Dioxane | 58 |
| 17 | I | 1.0 | Solvent-free | 93 |
As expected, the heterobimetallic dinuclear lanthanide/sodium alkoxide complexes (I–IV) could be used as catalysts for the synthesis of methyl benzylcarbamate (3aa), which was efficiently achieved with a yield ranging from 71% to 85% at 80 °C for 12 h with 1.0 mol% catalyst loading (Table 1, entries 2–5). In contrast, the desired reaction was conducted with very low reactivity and afforded only 8% yield of 3aa without a catalyst (Table 1, entry 1). Moreover, the monometallic lanthanide complex Nd(OCH2CH2NMe2)3, as well as the sodium alkoxide complex NaOCH2CH2NMe2, showed much lower catalytic activity, and the corresponding 3aa was obtained with only 18% and 49% yields even by 2% and 8% catalyst loadings (Table 1, entries 6 and 7). Generally, monometallic lanthanide alkoxide complex can be regarded as a soft Lewis acid catalyst, and monometallic sodium alkoxide complex may be used as a hard Brønsted base catalyst. The experimental results indicated that heterobimetallic lanthanide/sodium alkoxide complexes can be treated as the cooperative acid–base bifunctional catalysts, which have evidently higher activity than that of monometallic ones as the simple acid or base catalyst. To further study the influence of nuclearity on the catalytic activity,20 the mononuclear lanthanide/sodium alkoxide analogues with well-defined X-ray crystal structures were used as the catalysts for this reaction. The results showed mononuclear catalysts obtained relatively lower catalytic activity than corresponding dinuclear catalysts with the same rare earth metal center Nd and Yb (Table 1, entries 2, 4, 8 and 9), and yields of 3aa catalyzed by mono- and dinuclear catalysts is 66% vs. 85% and 54% vs. 71%, respectively. On the premise of having similar alkali metal alkoxide structures, the less Lewis acid center may lead to the relatively poor performance of mononuclear catalysts. Furthermore, it is found that the reactivity depends deeply on the lanthanide metal of the complex. The activity sequence observed here (Table 1, entries 2–5, 8 and 9) is consistent with the increase in ionic radius (Y ≈ Yb < Sm < Nd). The increasing of catalyst loading from 0.5% to 2.0% led to greater yield from 76% to 90% (Table 1, entries 2 and 10–12). Different solvents were screened in order to increase the conversion in the model reaction catalyzed by I at 80 °C with 1.0 mol% catalyst loading (Table 1, entries 2 and 13–16). Aromatic hydrocarbon solvent toluene was found to be a better reaction medium than polar aprotic (DMF, DMSO, CH3CN and dioxane) solvents. Polar aprotic solvents inhibited the reaction to some degree potentially due to coordination of the solvent to lanthanide center, which diminished the Lewis acidity of the catalyst. Considering several methodologies used excess amounts of carbonates (5 or even 10 equiv.) as a solvent, our research attempts to obtain an economic and green synthesis method without excess dialkyl carbonates and other solvents. It's satisfied that the reaction could perform well without excess substrates and other solvents under the present catalyst system and the best results were that 93% yield of carbamate 3aa was achieved at 80 °C with 1.0 mol% catalyst loading for 12 h under solvent-free conditions when the reagent component ratio of 1a to 2a is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 17).
:1 (Table 1, entry 17).
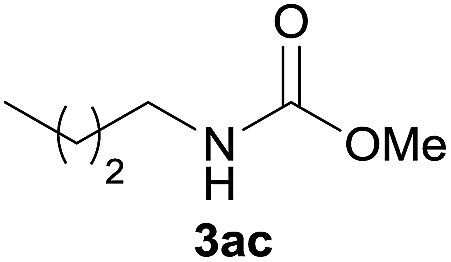
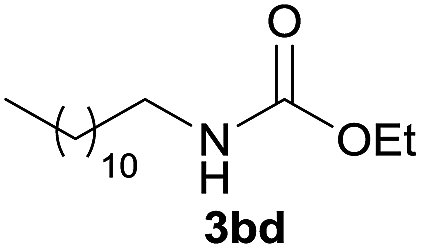
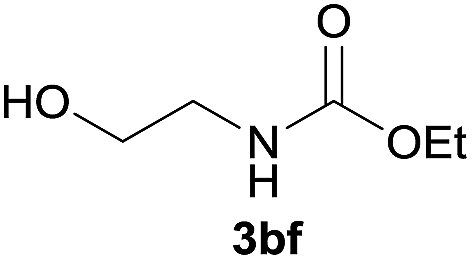
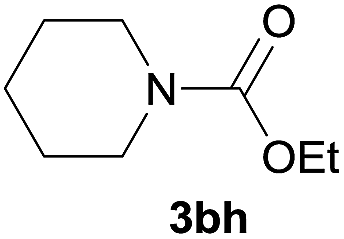
Under the optimized reaction conditions, a series of amines reacted with DMC and the results are presented in Table 2. From Table 2, it is obvious that transformation of various aliphatic primary and secondary amines with DMC could proceed to give the corresponding carbamates with moderate to high yields. Aliphatic primary amines such as n-butylamine (2c) and n-dodecylamine (2d) were effectively transformed with high yields up to 96% and 94% (Table 2, entries 3 and 4). 3-(Trimethoxysilyl) propan-1-amine (2e) known as silane coupling agent KH540, reacted smoothly with DMC to produce corresponding carbamate (3ae) with 90% yield (Table 2, entry 5). Meanwhile, aliphatic secondary amines 2h, 2i and 2j were efficiently catalyzed by I, the corresponding products also have good yields (Table 2, entries 8–10). It is found that both the electronic nature and steric factor had remarkable effect on the yield of this reaction. For 2-picolylamine (2b), because pyridine is a weak electron withdrawing group compared with phenyl group, the yield of 3ab is 89% (Table 2, entry 2), which is lower than that of 3aa (93%). Meanwhile, since the p–π conjugated effect of aromatic amine reduced the nucleophilicity of amine group, the reaction with aromatic amines proceeded less smoothly and lower yields was achieved under the same conditions. Aniline and substituted anilines, however, did not proceed resulted from the lower nucleophilicity. Moreover, highly selective amidation, not the possible transesterification, proceeded when DMC treated with ethanolamine (2f) (Table 2, entry 6). In addition, the double transformation of diamine with excess DMC was also successfully observed (Table 2, entry 7). Although diethyl carbonate (DEC) could react smoothly with corresponding amines compared with dimethyl carbonate (DME), the yields were relatively lower with the reaction time extending to 24 h (Table 2, entries 11–14).
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yieldb (%) |
| a Reaction conditions: carbonate 1 (2 mmol), amine 2 (2 mmol) and I (0.02 mmol) at 80 °C for 12 h under an argon atmosphere and solvent-free conditions.b Isolated yield.c Dimethyl carbonate 1a (4.2 mmol), amine 2 (2 mmol) and I (0.02 mmol).d Diethyl carbonate (DEC) 1b (2 mmol), amine 2 (2 mmol) and I (0.02 mmol) for 24 h. | |||
| 1 |  |
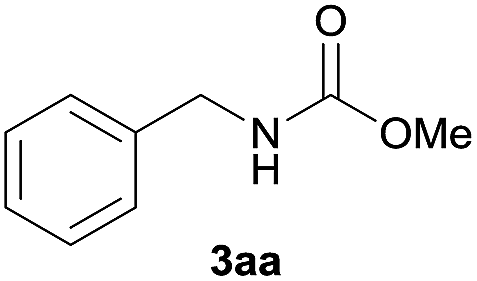 |
93 |
| 2 | 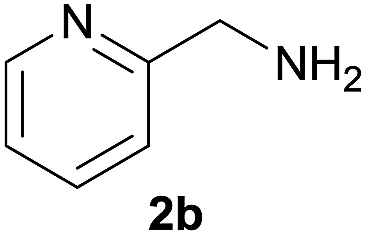 |
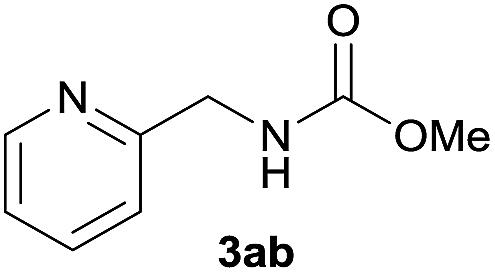 |
89 |
| 3 | 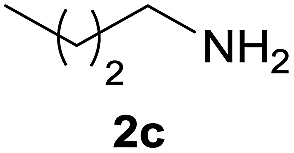 |
 |
96 |
| 4 | 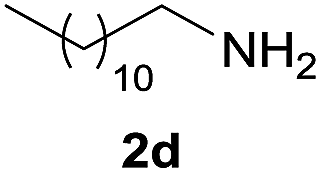 |
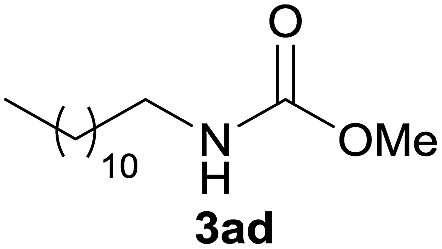 |
94 |
| 5 | 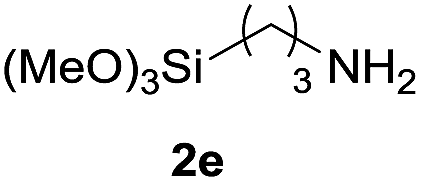 |
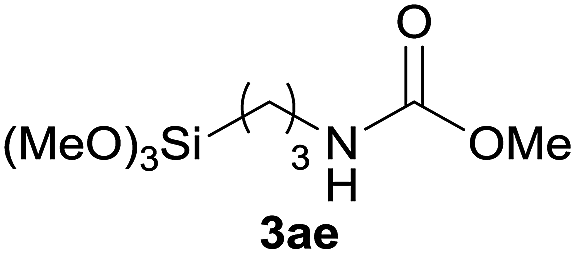 |
90 |
| 6 | 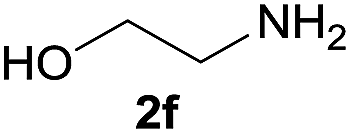 |
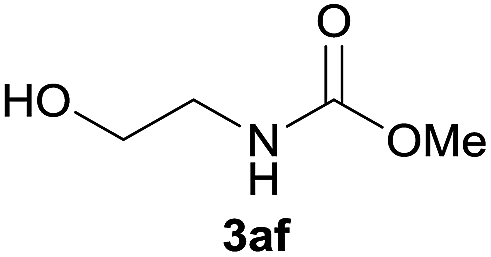 |
91 |
| 7c |  |
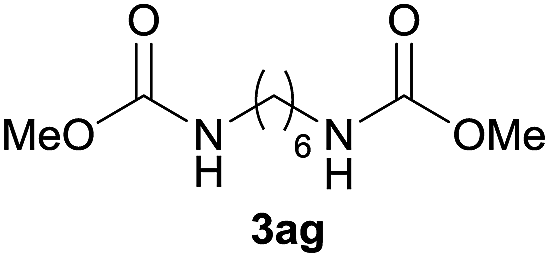 |
90 |
| 8 | 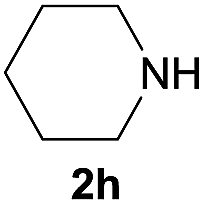 |
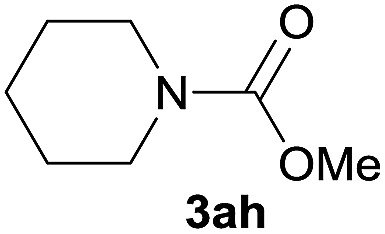 |
88 |
| 9 | 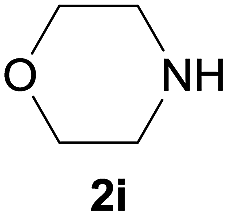 |
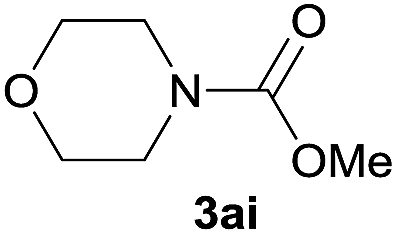 |
84 |
| 10 | 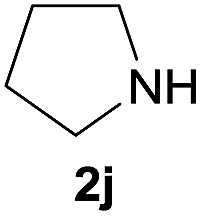 |
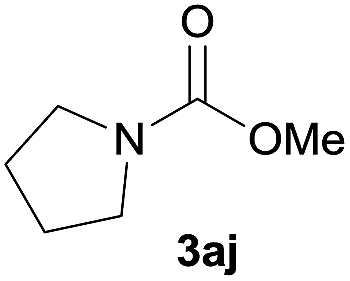 |
87 |
| 11d |  |
 |
76 |
| 12d | 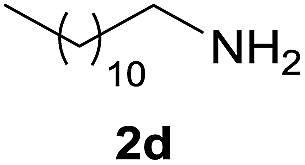 |
 |
79 |
| 13d | 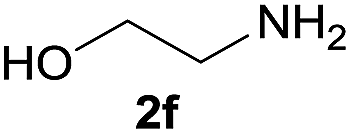 |
 |
72 |
| 14d | 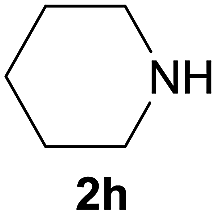 |
 |
72 |
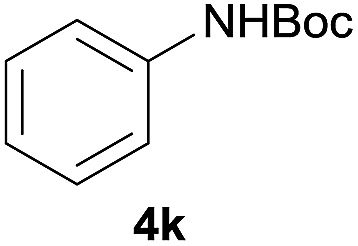
Carbamates are usually used as intermediates to protect the functionality of amines in the multistep organic synthesis.21 Among the protecting groups for amines, the tert-butoxycarbonyl (Boc) is one of the most used, owing to the stability of N-tert-butylcarbamates toward nucleophiles or strong basic conditions and catalytic hydrogenation.22 Various methods have been used to protect amines in their N-Boc form over the years.23 These methods, however, have several drawbacks such as long reaction time, low yields, usage of solvents, formation of side-products and complicated reaction procedures, etc. In order to better standardize the reaction, different catalyst loadings of I were used for the N-Boc protection of aniline at room temperature under solvent-free conditions. The results are shown in Table 3. It was found that the reaction proceeded with only 54% yield until 240 min without a catalyst (Table 3, entry 1). And it is clear that the yield increased from 64% to 91% on varying the loading of catalyst from 0.1 mol% to 1.5 mol%. The best results were that 89% yield of product was achieved within 10 min at room temperature with 0.5 mol% catalyst loading under solvent-free conditions when the molar ratio of (Boc)2O to anilineis 1:1 (Table 3, entry 4).
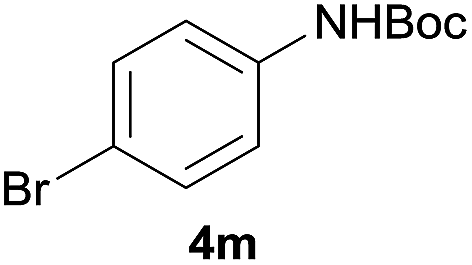
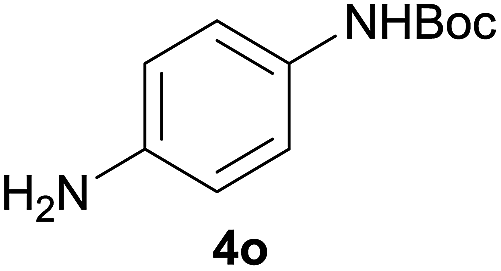
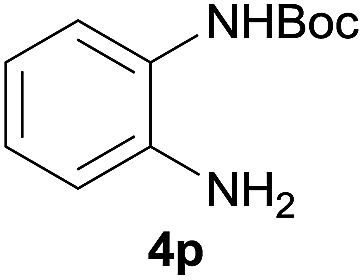
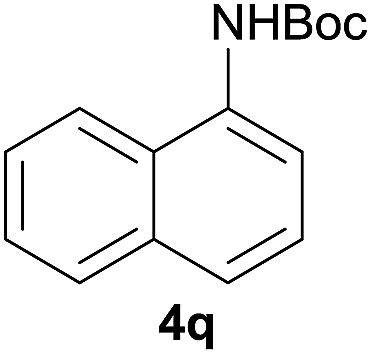
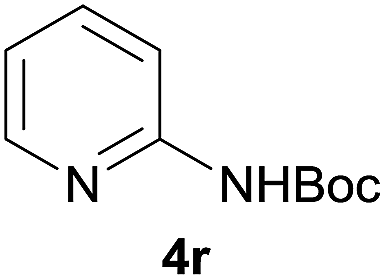
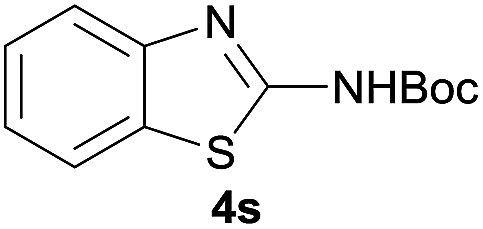
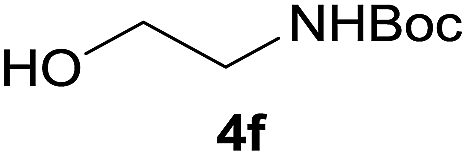
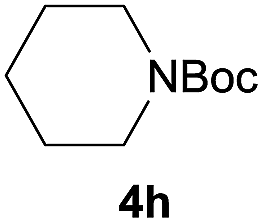
After optimization of the reaction conditions, various aromatic, heteroaromatic and aliphatic amines were converted to the N-Boc derivatives under the selected conditions. Table 4 summarizes the results. Obviously, the catalyst system was very effective for the N-Boc protection of the amines. No competitive formation of N,N-di-BOC, urea and isocyanate was observed.24 Various derivatives of aniline were taken under the reaction conditions and the products were obtained with good yields. As expected, electron donating groups on aryl groups gave rise to higher yields than electron withdrawing groups (Table 4, entries 1–4). Substrates having an OH group afforded the N-Boc derivatives without O-Boc formation. Protection of diamines in the presence of I was also studied. It was found that selective mono protection of diamines was achieved using an equivalent of (Boc)2O, the mono-N-Boc-protected products were obtained with high yields (Table 4, entries 5 and 6). Moreover, the protocol could also work well with naphthylamine and heteroaromatic amines (Table 4, entries 7–9). Benzyl amine, 2-picolylamine and aliphatic secondary amines reacted more quickly than aniline derivatives due to their high nucleophilicity (Table 4, entries 10, 11 and 13–15). Furthermore, the chemoselectivity was further demonstrated in the case of ethanolamine that did not form O-Boc formation and afforded the corresponding N-Boc product with high yield (Table 4, entry 12).
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yieldb (%) |
| a Reaction conditions: amine (1 mmol), (Boc)2O (1 mmol) and I (0.5 mol%) at room temperature for 10 min under solvent-free conditions.b Isolated yield.c Reaction time: 1 h.d Reaction time: 30 min.e Reaction time: 2 min. | |||
| 1 | 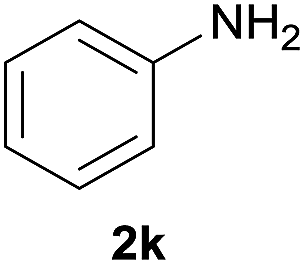 |
 |
87 |
| 2 |  |
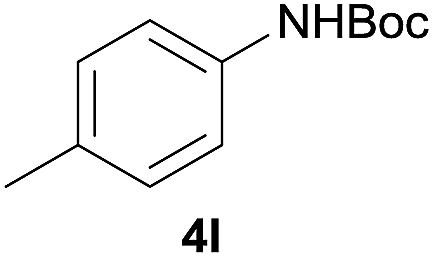 |
93 |
| 3 |  |
 |
91 |
| 4 |  |
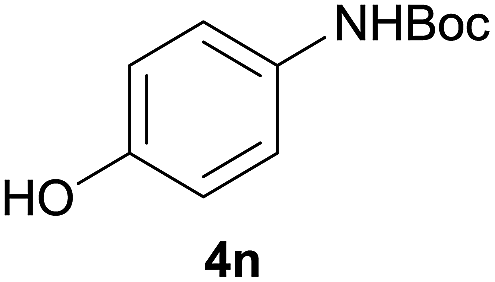 |
96 |
| 5 | 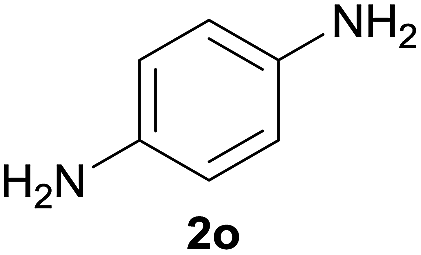 |
 |
95 |
| 6 | 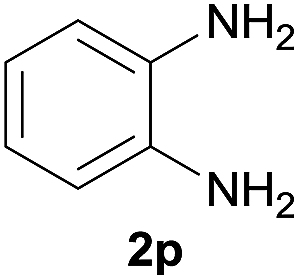 |
 |
93 |
| 7c | 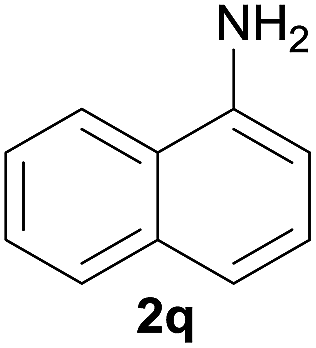 |
 |
95 |
| 8d | 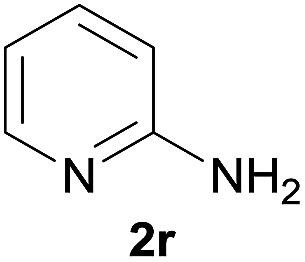 |
 |
92 |
| 9d | 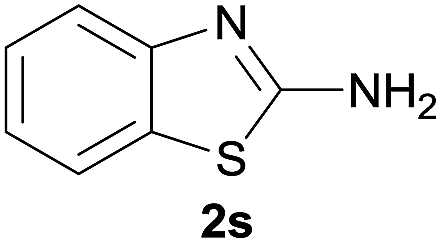 |
 |
90 |
| 10e | 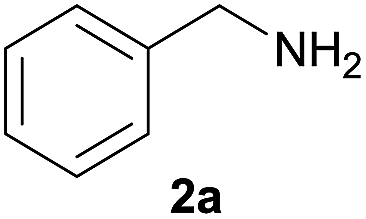 |
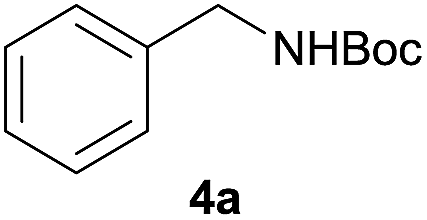 |
99 |
| 11e | 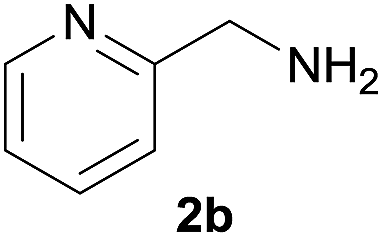 |
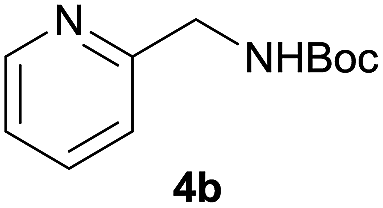 |
98 |
| 12 | 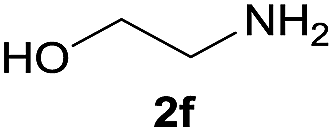 |
 |
96 |
| 13e | 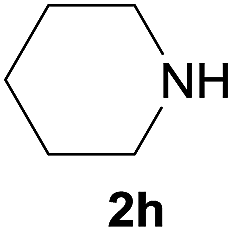 |
 |
98 |
| 14e | 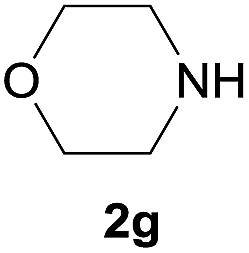 |
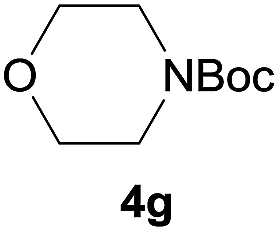 |
98 |
| 15e | 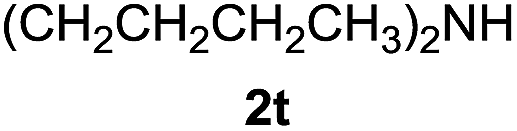 |
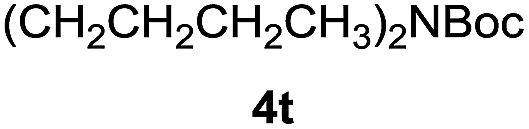 |
98 |
The bimetallic cooperative catalysts could utilize many different types of metals, such as alkali metals, transition metals and lanthanides.25 The important role of lanthanide in the bimetallic catalysts, was studied by P. J. Walsh etc. groups from X-ray and NMR analysis.26 Heterobimetallic dinuclear lanthanide alkoxide complexes have stronger activation than that of mononuclear analogues due to the double Lewis acid center. With consideration of these results and other mechanism studies of acid–base bifunctional catalysis in the literature,14,27 a plausible mechanism of the reaction of carbonates and amines is illustrated in Scheme 1. Firstly, heterobimetallic dinuclear lanthanide/sodium alkoxide complex I activates both the carbonate and amine to generate the adduct A, wherein two lanthanide metal ions work as double Lewis acid centers for activating simultaneously the carbonyl and alkoxy oxygen atoms of carbonate, while the sodium alkoxide moiety functions as a Brønsted base to bring the nucleophile amine in close proximity. Then the formation of a sp3 hybridized tetrahedral intermediate (B) followed by the nucleophilic attack of the activated amine on the electrophilic carbon center of the carbonyl moiety. Subsequently, the sterically congested B collapses to regenerate the sp2 hybridized carbonyl unit and the active species I, resurrected along with the release of product carbamate and alcohol.
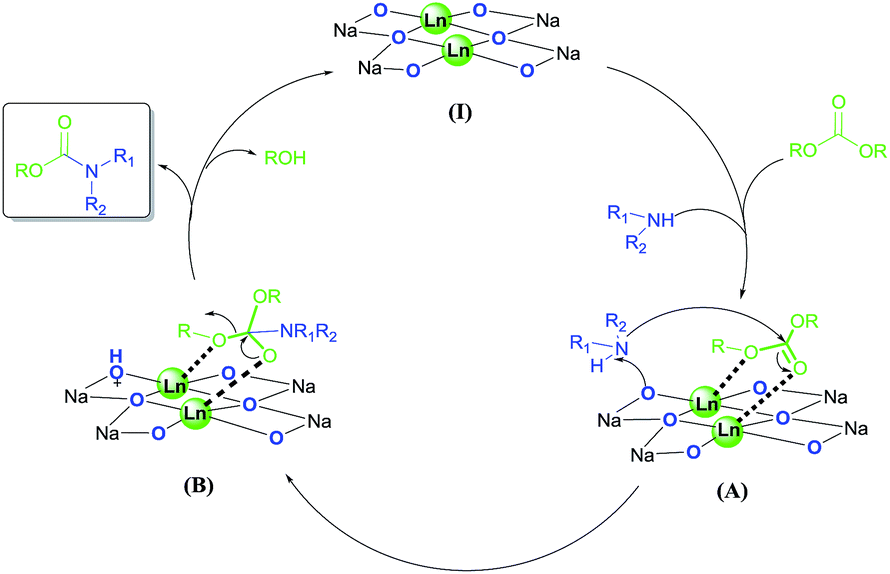 | ||
| Scheme 1 Proposed catalytic cycle for synthesis of carbamates from carbonates with amines. | ||
Conclusions
In summary, heterobimetallic dinuclear lanthanide alkoxide complexes were used as a new class of acid–base bifunctional catalysts for synthesis of carbamates from DMC/DEC with amines as well as the N-Boc protection of various amines. The new catalysts showed high catalytic activity and a wide scope of substrates with good to excellent yields under solvent-free conditions. In this catalyst design, two lanthanide metals of dinuclear catalysts act as the binary Lewis acid centers to activate electrophiles, while alkali metal alkoxides serve as a Brønsted base to bring nucleophiles in close proximity.Experimental
General methods
All manipulations and reactions were performed under an atmosphere of argon with standard Schlenk techniques. The carbonates and amines were obtained commercially. Heterobimetallic lanthanide/sodium alkoxide complexes Ln2Na8(OCH2CH2NMe2)12(OH)2 [Ln: I (Nd), II (Sm), III (Yb) and IV (Y)], LnNa8(OtBu)10(OH) [Ln: i (Nd) and ii (Yb)] and the corresponding monometallic complex Nd(OCH2CH2NMe2)3 were prepared according to the literature.18a,d 1H and 13C NMR spectra were recorded with Bruker Avance 400 MHz spectrometer using CDCl3 as the solvent with tetramethylsilane (TMS) as the internal standard. The GC (Shimadzu GC-2010plus) and GCMS (Shimadzu GCMS-QP2010) analyses were carried out using N2 and He as the carrier gas, respectively. High-resolution mass spectra (HRMS) were carried out using a TOF-MS instrument with an EI or ESI source. Column chromatography was performed using silica gel (100–200 mesh). Petroleum ether (PE) used was the fraction boiling in the range 60–90 °C.Typical procedure for the synthesis of carbamates from amines and dialkyl carbonates (3aa as an example)
A mixture of Nd2Na8(OCH2CH2NMe2)12(OH)2 (I) (0.02 mmol), dimethyl carbonate (2 mmol) and benzyl amine (2 mmol) in a 10 mL Schlenk tube was stirred at 80 °C for the 12 h under argon. The resulting mixture was then filtered through a small plug of silica gel to remove the catalyst. The crude product was purified by silica gel column chromatography (EtOAc/PE = 1:4) to provide the title compound (307 mg, 93% yield) as a white solid.
Typical procedure for the N-Boc protection of amines (4k as an example)
A typical procedure for the N-Boc protection of amines from (Boc)2O and aniline is as follows: in a 10 mL Schlenk tube with Nd2Na8(OCH2CH2NMe2)12(OH)2 (I) (0.005 mmol), (Boc)2O (1 mmol) and aniline (1 mmol), the mixture was then stirred at room temperature for 10 min. The resulting mixture was then filtered through a small plug of silica gel to remove the catalyst. The crude product was purified by silica gel column chromatography (EtOAc/PE = 1:6) to provide the title compound (168 mg, 87% yield) as a white solid.
Analysis data for products
Acknowledgements
We acknowledge financial support by the National Natural Science Foundation of China (No. 20902001, 21102001), the Natural Science Foundation of Education Department of Anhui Province (No. KJ2014A013, KJ2015A047), Anhui Provincial Natural Science Foundation (1608085MB39), and the 211 Project of Anhui University.Notes and references
- (a) T. T. Wu, J. Huang, N. D. Arrington and G. M. Dill, J. Agric. Food Chem., 1987, 35, 817–823 CrossRef CAS; (b) A. J. Wills, Y. Krishnan-Ghosh and S. Balasubramanian, J. Org. Chem., 2002, 67, 6646–6652 CrossRef CAS PubMed; (c) S. Ray and D. Chaturvedi, Drugs Future, 2004, 29, 343–357 CrossRef CAS; (d) T. Goto, Y. Ito, S. Yamada, H. Matsumoto, H. Oka and H. Nagase, Anal. Chim. Acta, 2006, 555, 225–232 CrossRef CAS; (e) J. Song, H. W. Shih and L. Deng, Org. Lett., 2007, 9, 603–606 CrossRef CAS PubMed; (f) E. M. Dangerfield, M. S. Timmer and B. L. Stocker, Org. Lett., 2009, 11, 535–538 CrossRef CAS PubMed; (g) V. Janganati, N. R. Penthala, N. R. Madadi, Z. Chen and P. A. Crooks, Bioorg. Med. Chem. Lett., 2014, 24, 3499–3502 CrossRef CAS PubMed; (h) I. B. Dhouib, A. Annabi, M. Jallouli, S. Elfazaa and M. M. Lasram, J. Appl. Biomed., 2016, 14, 85–90 CrossRef.
- (a) H. Eckert and B. Forster, Angew. Chem., Int. Ed., 1987, 26, 894–895 CrossRef; (b) J. S. Nowick, N. A. Powell, T. M. Nguyen and G. Noronha, J. Org. Chem., 1992, 57, 7364–7366 CrossRef CAS; (c) P. Majer and R. S. Randad, J. Org. Chem., 1994, 59, 1937–1938 CrossRef CAS.
- (a) R. N. Salvatore, J. A. Ledger and K. W. Jung, Tetrahedron Lett., 2001, 42, 6023–6025 CrossRef CAS; (b) F. Shi, J. J. Peng and Y. Q. Deng, J. Catal., 2003, 219, 372–375 CrossRef CAS.
- (a) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed; (b) F. Ragaini, C. Cognolato, M. Gasperini and S. Cenini, Angew. Chem., Int. Ed., 2003, 42, 2886–2889 CrossRef CAS PubMed; (c) F. Shi, Y. D. He, D. M. Li, Y. B. Ma, Q. H. Zhang and Y. Q. Deng, J. Mol. Catal. A: Chem., 2006, 244, 64–67 CrossRef CAS.
- (a) M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 151–152 RSC; (b) R. N. Salvatore, S. I. Shin, A. S. Nagle and K. W. Jung, J. Org. Chem., 2001, 66, 1035–1037 CrossRef CAS PubMed; (c) D. L. Kong, L. N. He and J. Q. Wang, Synlett, 2010, 1276–1280 CAS; (d) H. L. An, L. L. Zhang and X. Q. Zhao, Chin. J. Chem. Eng., 2014, 22, 607–610 CrossRef CAS; (e) W. F. Xiong, C. R. Qi, H. T. He and H. F. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3084–3087 CrossRef CAS PubMed; (f) W. F. Xiong, C. R. Qi, Y. B. Peng and H. F. Jiang, Chem.–Eur. J., 2015, 21, 14314–14318 CrossRef CAS PubMed.
- A. R. Sardarian and I. D. Inaloo, RSC Adv., 2015, 5, 76626–76641 RSC.
- S. Y. Moon, U. B. Kim and W. S. Kim, J. Org. Chem., 2015, 80, 1856–1865 CrossRef CAS PubMed.
- (a) P. Gogoi and D. Konwar, Tetrahedron Lett., 2007, 48, 531–533 CrossRef CAS; (b) A. R. Modarresi-Alam, F. Khamooshi, M. Nasrollahzadeh and H. A. Amirazizi, Tetrahedron, 2007, 63, 8723–8726 CrossRef CAS.
- (a) P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195–3200 RSC; (b) R. K. Sharma, S. Dutta and S. Sharma, Dalton Trans., 2015, 44, 1303–1316 RSC.
- (a) T. Baba, M. Fujiwara, A. Oosaku, A. Kobayashi, R. G. Deleon and Y. Ono, Appl. Catal., A, 2002, 227, 1–6 CrossRef CAS; (b) M. Curini, F. Epifano, F. Maltese and O. Rosati, Tetrahedron Lett., 2002, 43, 4895–4897 CrossRef CAS; (c) F. Li, W. B. Li, J. Li, W. Xue, Y. J. Wang and X. Q. Zhao, Appl. Catal., A, 2014, 475, 355–362 CrossRef CAS; (d) V. Mishra, J. K. Cho, S. H. Shin, Y. W. Suh and Y. J. Kim, Appl. Catal., A, 2014, 487, 82–90 CrossRef CAS.
- (a) S. P. Gupte, A. B. Shivarkar and R. V. Chaudhari, Chem. Commun., 2001, 2620–2621 RSC; (b) S. Carloni, D. E. De Vos, P. A. Jacobs, R. Maggi, G. Sartori and R. Sartorio, J. Catal., 2002, 205, 199–204 CrossRef CAS; (c) M. Selva, P. Tundo and A. Perosa, Tetrahedron Lett., 2002, 43, 1217–1219 CrossRef CAS; (d) T. Sima, S. Guo, F. Shi and Y. Deng, Tetrahedron Lett., 2002, 43, 8145–8147 CrossRef CAS; (e) M. Selva, P. Tundo, A. Perosa and F. Dall'Acqua, J. Org. Chem., 2005, 70, 2771–2777 CrossRef CAS PubMed; (f) C. Han and J. A. Porco Jr, Org. Lett., 2007, 9, 1517–1520 CrossRef CAS PubMed; (g) H. Zhou, F. Shi, X. Tian, Q. Zhang and Y. Deng, J. Mol. Catal. A: Chem., 2007, 271, 89–92 CrossRef CAS; (h) N. Lucas, A. P. Amrute, K. Palraj, G. V. Shanbhag, A. Vinu and S. B. Halligudi, J. Mol. Catal. A: Chem., 2008, 295, 29–33 CrossRef CAS; (i) B. Wang, J. He and R. C. Sun, Chin. Chem. Lett., 2010, 21, 794–797 CrossRef CAS; (j) S. Kumar and S. L. Jain, New J. Chem., 2013, 37, 2935–2938 RSC.
- (a) G. K. Prakash, T. Mathew and G. A. Olah, Acc. Chem. Res., 2012, 45, 565–577 CrossRef CAS PubMed; (b) K. Shen, X. H. Liu, L. L. Lin and X. M. Feng, Chem. Sci., 2012, 3, 327–334 RSC; (c) D. J. Averill and M. J. Allen, Catal. Sci. Technol., 2014, 4, 4129–4137 RSC; (d) J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS PubMed; (e) X. Gu, X. Zhu, Y. Wei, S. Wang, S. Zhou, G. Zhang and X. Mu, Organometallics, 2014, 33, 2372–2379 CrossRef CAS; (f) J. Qin, P. Wang, Q. Li, Y. Zhang, D. Yuan and Y. Yao, Chem. Commun., 2014, 50, 10952–10955 RSC; (g) J. R. Robinson, J. Yadav, X. Y. Fan, G. R. Stanton, E. J. Schelter, M. A. Pericas and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 1243–1254 CrossRef CAS; (h) X. Wang, S. Y. Li, Y. M. Pan, H. S. Wang, H. Liang, Z. F. Chen and X. H. Qin, Org. Lett., 2014, 16, 580–583 CrossRef CAS PubMed; (i) P.-H. Wei, L. Xu, L.-C. Song, W.-X. Zhang and Z. Xi, Organometallics, 2014, 33, 2784–2789 CrossRef CAS; (j) B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102–1117 RSC; (k) Q. F. Xing, T. Zhao, S. Z. Du, W. H. Yang, T. L. Liang, C. Redshaw and W. H. Sun, Organometallics, 2014, 33, 1382–1388 CrossRef CAS.
- (a) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed; (b) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed; (c) M. Shibasaki and S. Matsunaga, J. Synth. Org. Chem., Jpn., 2010, 68, 1142–1149 CrossRef CAS; (d) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2013, 52, 223–234 CrossRef CAS PubMed.
- (a) H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194–6198 CrossRef CAS; (b) M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed., 1997, 36, 1236–1256 CrossRef; (c) Y. M. A. Yamada, N. Yoshikawa, H. Sasai and M. Shibasaki, Angew. Chem., Int. Ed., 1997, 36, 1871–1873 CrossRef CAS; (d) E. Emori, T. Arai, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 4043–4044 CrossRef CAS; (e) K.-I. Yamada, S. J. Harwood, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 3504–3506 CrossRef CAS.
- S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- (a) S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900–4901 CrossRef CAS PubMed; (b) S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 4925–4934 CrossRef CAS PubMed.
- (a) T. Nitabaru, A. Nojiri, M. Kobayashi, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 13860–13869 CrossRef CAS PubMed; (b) D. Sureshkumar, K. Hashimoto, N. Kumagai and M. Shibasaki, J. Org. Chem., 2013, 78, 11494–11500 CrossRef CAS PubMed.
- (a) H. Sheng, F. Xu, Y. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 7722–7724 CrossRef CAS PubMed; (b) H. T. Sheng, L. Y. Zhou, Y. Zhang, Y. M. Yao and Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1210–1218 CrossRef CAS; (c) H.-T. Sheng, J.-M. Li, Y. Zhang, Y.-M. Yao and Q. Shen, Polyhedron, 2008, 27, 1665–1672 CrossRef CAS; (d) H. T. Sheng, J. M. Li, Y. Zhang, Y. M. Yao and Q. Shen, J. Appl. Polym. Sci., 2009, 112, 454–460 CrossRef CAS; (e) H. T. Sheng, Y. Feng, Y. Zhang and Q. Shen, Eur. J. Inorg. Chem., 2010, 5579–5586 CrossRef CAS; (f) H. Sheng, J. Shi, Y. Feng, H. Wang, Y. Jiao, H. Sheng, Y. Zhang and Q. Shen, Dalton Trans., 2012, 41, 9232–9240 RSC; (g) R. Zeng, H. Sheng, Y. Zhang, Y. Feng, Z. Chen, J. Wang, M. Chen, M. Zhu and Q. Guo, J. Org. Chem., 2014, 79, 9246–9252 CrossRef CAS PubMed.
- K. Agura, Y. Hayashi, K. Mashima and T. Ohshima, Chem.–Asian J., 2016, 11, 1548–1554 CrossRef CAS PubMed.
- (a) L. Li, M. V. Metz, H. Li, M. C. Chen, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12725–12741 CrossRef CAS PubMed; (b) M. Yamamura, H. Miyazaki, M. Iida, S. Akine and T. Nabeshima, Inorg. Chem., 2011, 50, 5315–5317 CrossRef CAS PubMed; (c) A. Abhervé, J. M. Clemente-Juan, M. Clemente-León, E. Coronado, J. Boonmak and S. Youngme, New J. Chem., 2014, 38, 2105–2113 RSC; (d) D. C. Charalampou, N. Kourkoumelis, S. Karanestora, L. P. Hadjiarapoglou, V. Dokorou, S. Skoulika, A. Owczarzak, M. Kubicki and S. K. Hadjikakou, Inorg. Chem., 2014, 53, 8322–8333 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, in Protective group inOrganic Synthesis, Wiley-VCH, New York, 4th edn, 2007 Search PubMed.
- G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS PubMed.
- (a) G. V. M. Sharma, J. Janardhan Reddy, P. Sree Lakshmi and P. Radha Krishna, Tetrahedron Lett., 2004, 45, 6963–6965 CrossRef CAS; (b) N. Suryakiran, P. Prabhakar, T. S. Reddy, K. Rajesh and Y. Venkateswarlu, Tetrahedron Lett., 2006, 47, 8039–8042 CrossRef CAS; (c) R. Varala, S. Nuvula and S. R. Adapa, J. Org. Chem., 2006, 71, 8283–8286 CrossRef CAS PubMed; (d) K. S. Kumar, J. Iqbal and M. Pal, Tetrahedron Lett., 2009, 50, 6244–6246 CrossRef CAS; (e) F. Jahani, M. Tajbakhsh, H. Golchoubian and S. Khaksar, Tetrahedron Lett., 2011, 52, 1260–1264 CrossRef CAS; (f) N. G. Khaligh, RSC Adv., 2012, 2, 12364–12370 RSC; (g) S. V. Atghia and S. SarviBeigbaghlou, J. Org. Chem., 2013, 745–746, 42–49 CrossRef CAS; (h) A. Chinnappan, D. La and H. Kim, RSC Adv., 2013, 3, 13324–13328 RSC; (i) S. Karimian and H. Tajik, Chin. Chem. Lett., 2014, 25, 218–220 CrossRef CAS.
- Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368–6380 CrossRef CAS PubMed.
- J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC.
- (a) H. C. Aspinall, J. F. Bickley, J. L. M. Dwyer, N. Greeves, R. V. Kelly and A. Steiner, Organometallics, 2000, 19, 5416–5423 CrossRef CAS; (b) L. Di Bari, M. Lelli, G. Pintacuda, G. Pescitelli, F. Marchetti and P. Salvadori, J. Am. Chem. Soc., 2003, 125, 5549–5558 CrossRef CAS PubMed; (c) A. J. Wooten, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2006, 45, 2549–2552 CrossRef CAS PubMed; (d) A. J. Wooten, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2008, 130, 7407–7419 CrossRef CAS PubMed.
- K. Ishihara, A. Sakakura and M. Hatano, Synlett, 2007, 686–703 CrossRef CAS.
- Z.-H. Guan, H. Lei, M. Chen, Z.-H. Ren, Y. Bai and Y.-Y. Wang, Adv. Synth. Catal., 2012, 354, 489–496 CrossRef CAS.
- Z. D. Crane, P. J. Nichols, T. Sammakia and P. J. Stengel, J. Org. Chem., 2011, 76, 277–280 CrossRef CAS PubMed.
- X. G. Guo, J. P. Shang, J. A. Li, L. G. Wang, Y. B. Ma, F. Shi and Y. Q. Deng, Synth. Commun., 2011, 41, 1102–1111 CrossRef CAS.
- M. Distaso and E. Quaranta, Tetrahedron, 2004, 60, 1531–1539 CrossRef CAS.
- M. Vargas-Sanchez, S. Lakhdar, F. Couty and G. Evano, Org. Lett., 2006, 8, 5501–5504 CrossRef CAS PubMed.
- J. E. Taylor, M. D. Jones, J. M. Williams and S. D. Bull, J. Org. Chem., 2012, 77, 2808–2818 CrossRef CAS PubMed.
- F. Szurdoki, A. Székács, H. M. Le and B. D. Hammock, J. Agric. Food Chem., 2002, 50, 29–40 CrossRef CAS PubMed.
- J. F. King, Z. R. Guo and D. F. Klassen, J. Org. Chem., 1994, 59, 1095–1101 CrossRef CAS.
- S. Okumura, Y. Takeda, K. Kiyokawa and S. Minakata, Chem. Commun., 2013, 49, 9266–9268 RSC.
- P. Gopinath, S. Nilaya and K. M. Muraleedharan, Org. Lett., 2011, 13, 1932–1935 CrossRef CAS PubMed.
- G. Barker, P. O'Brien and K. R. Campos, Org. Lett., 2010, 12, 4176–4179 CrossRef CAS PubMed.
- S. K. Verma, R. Ghorpade, A. Pratap and M. P. Kaushik, Tetrahedron Lett., 2012, 53, 2373–2376 CrossRef CAS.
- G. A. Molander and I. Shin, Org. Lett., 2013, 15, 2534–2537 CrossRef CAS PubMed.
- C. Stock and R. Bruckner, Adv. Synth. Catal., 2012, 354, 2309–2330 CrossRef CAS.
- F. Ma, X. Xie, L. Zhang, Z. Peng, L. Ding, L. Fu and Z. Zhang, J. Org. Chem., 2012, 77, 5279–5285 CrossRef CAS PubMed.
- F. Jahani, M. Tajbakhsh, H. Golchoubian and S. Khaksar, Tetrahedron Lett., 2011, 52, 1260–1264 CrossRef CAS.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed.
- F. Troisi, A. Russo, C. Gaeta, G. Bifulco and P. Neri, Tetrahedron Lett., 2007, 48, 7986–7989 CrossRef CAS.
- C. T. Chen, J. H. Kuo, V. D. Pawar, Y. S. Munot, S. S. Weng, C. H. Ku and C. Y. Liu, J. Org. Chem., 2005, 70, 1188–1197 CrossRef CAS PubMed.
- T. Vilaivan, Tetrahedron Lett., 2006, 47, 6739–6742 CrossRef CAS.
- E. Alonso, D. J. Ramon and M. Yus, Tetrahedron, 1997, 53, 14355–14368 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for the products. See DOI: 10.1039/c6ra15160d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |