
Exploring saccharinate-tetrazoles as selective Cu(ii) ligands: structure, magnetic properties and cytotoxicity of copper(ii) complexes based on 5-(3-aminosaccharyl)-tetrazoles†
 ab
ab
Abstract
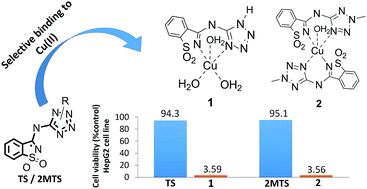
The role of copper in the proliferation of cancer cells is under investigation and has been explored in the context of cancer chemotherapy. The evidence that proliferation of cancer cells requires a higher abundance of Cu(II) than their normal counterparts has prompted the development of new copper chelators that can avidly bind copper ions, forming redox active metal complexes that ultimately lead to harmful reactive oxygen species (ROS) in neoplasms. In this context, the mandatory properties of the chelators for medical applications are safety (neglectable cytotoxicity), high binding affinity and selectivity towards Cu(II). We report the synthesis, structure (calculations and single crystal X-ray diffraction), spectroscopic (IR; UV-Vis) and magnetic properties of two novel copper(II) complexes based on 5-(3-aminosaccharyl)-tetrazoles (TS and 2MTS), as well as their in vitro cytotoxicity against the human hepatic carcinoma cell line HepG2. Quite interestingly, we found that the saccharinate-tetrazoles tested exhibit strong binding selectivity to Cu(II), over Fe(II) and Ca(II). Additionally, the corresponding copper complexes have shown a huge increase in the in vitro cytotoxicity against tumoral cells, compared to the corresponding nontoxic ligands. Thus, the new ligands may be viewed as potential precursors of selective cytotoxic agents, acting as non-cytotoxic pro-drugs that can be activated inside neoplastic cells, known to be richer in Cu(II) than the corresponding normal cells.
 Please wait while we load your content...
Please wait while we load your content...

