
Preparation of photoreactive nanocellulosic materials via benzophenone grafting†
Hannes Orelma‡
*ab,
Maija Vuoriluotoa,
Leena-Sisko Johanssona,
Joseph M. Campbella,
Ilari Filpponena,
Markus Biesalskib and
Orlando J. Rojasa
aBiobased Colloids and Materials (BiCMat), Aalto University, School of Chemical Technology, Department of Forest Products Technology, 00076 Aalto, PL 11000, Espoo, Finland. E-mail: hannes.orelma@vtt.fi
bLaboratory of Macromolecular Chemistry and Paper Chemistry, Department of Chemistry, Technische Universitat Darmstadt, 64287 Darmstadt, Germany
First published on 2nd September 2016
Abstract
A method for preparing photo-crosslinkable cellulose nanofibrils (CNF) was investigated. Benzophenone (BP), a UV-radical crosslinker, was chemically grafted to TEMPO-oxidized wood fibers in water/DMSO medium. This resulted in a reduction of carboxyl group content together with an increase of the number density of amide linkages. The BP-functionalized fibres were then microfluidized into TEMPO-oxidized CNF (TOCNF). As evidence of the crosslinking performance, the films of BP-activated TOCNF displayed improved water resistance upon UV curing (even under high energy sonication). In addition to improving wet strength the method is suitable for further modification of CNF, either by utilizing the remaining free carboxyl groups or via photochemical grafting of other substances onto the CNF structure.
Introduction
Nanofibrillated cellulose (CNF), a young member of the lignocellulosic materials family, shares many of the interesting characteristics of the group, such as biocompatibility, biodegradability, and excellent stability.1,2 CNF also has a remarkably high gel forming ability, which can be utilised e.g. in film forming,3 functional aerogels,4 textile fibers,5,6 and as rheology modifiers.7 Therefore, CNF has significant potential in the development of high-tech biotechnological materials.When cellulosic materials are fibrillated into CNF, carboxyl groups play an important role in easing the disintegration process of fibrils by increasing swelling and inter fibrillar electrostatic repulsions, thus lowering demands in mechanical energy.8 Moreover, carboxyl groups are useful in gel formation and in grafting or conjugating functionalities onto CNF. However, high carboxyl content in CNF may compromise the wet strength of the material.
Wet strength of CNF may be improved by addition of cationic or non-anionic polymers.9,10 However, other routes may be desirable when bi-functional CNF-materials are required, for example if carboxyl (or other charged) groups are needed for activation in addition to e.g. water resistance. Light-induced crosslinking is a relatively new approach for improving the wet strength of cellulosic materials, especially if achieved without addition of chemical additives. For example benzophenone (BP) can provide radical-based crosslinking under UV irradiation.11 Benzophenone grafted polymers have been found to work effective with polymeric thin films in grafting polymeric materials and internally cross-linked polymeric layers12,13 and BP is widely utilized for UV protection of plastics, soaps, and perfumes.
Under UV-exposure, benzophenone photolyse to a highly reactive triplet state, a ketone intermediate via n–π* or π–π* transition, depending on the UV-light wave length.14 Benzophenone radicals can react with hydrogens from aliphatic CH groups via H-abstraction/recombination mechanisms. Moreover, unreacted BP molecules can undergo multiple photolytic activation cycles improving the conjugation efficiency. The conjugation of BP onto cellulose has been recently introduced for UV-crosslinking cellulose nanocrystal (CNC) films, by covalently grafting benzophenone on surfaces of CNC.15 Moreover, benzophenone has been used with functional paper devices as an UV-reactive crosslinker by incorporating benzophenone into methyl methacrylate polymer chain.16,17 When a benzophenone molecule is grafted onto cellulose, under UV-exposure the radicalized benzophenone substitute may abstract hydrogen from both C1-carbon and C6-ketone (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of a cellulose glucopyranose ring located to the neighbouring cellulose chain leading to a covalent bond formation. When benzophenone is located onto CNF fibril, fibrils carrying benzophenone substituents may covalently attach to neighbouring fibrils unsubstituted spots.
O) of a cellulose glucopyranose ring located to the neighbouring cellulose chain leading to a covalent bond formation. When benzophenone is located onto CNF fibril, fibrils carrying benzophenone substituents may covalently attach to neighbouring fibrils unsubstituted spots.
In this work, BP was covalently grafted onto CNF using a tailored sequential pre-functionalization strategy. First the precursor wood fibers were TEMPO-oxidized to open their structure. Then BP was grafted onto the carboxylic groups via aqueous EDC/NHS coupling (Scheme 1) and only after functionalisation the fibers were disintegrated into cellulosic nanofibrils (TEMPO-oxidized CNF or TOCNF) using high pressure fluidization. Prepared TOCNF (BP-TOCNF) was cast into films and crosslinked using UV light. The method presented herein is not only suitable for improving the wet strength of CNF but can also potentially be utilized for the photochemical grafting of various monomeric or polymeric additives onto the CNF backbone. This developed strategy is proposed for greener nanolignocellulosic materials.
 | ||
| Scheme 1 Schematic illustration on the manufacture of benzophenone functionalized nanocellulose (BP-TOCNF) materials. | ||
Experimental section
TEMPO ((2,2,6,6-tetramethyl-piperidin-1-yl)oxyl, #426369) free radical, 4-aminobenzophenone (#A41402), NHS (N-hydroxysuccinimide, #130672), EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride, #03450), were obtained from Sigma-Aldrich (Helsinki, Finland). The water used in all experiments was deionized and further purified with a Millipore Synergy UV unit (MilliQ-water). All chemicals were used without any further purification.Fiber carboxylation
Bleached birch fibers were carboxylated with alkaline 2,2,6,6,-tetramethylpipelidine-1-oxyl radical (TEMPO)–NaBr–NaClO system as described by Isogai et al.18 Briefly, 0.9 g TEMPO (1.3 mmol l−1) and 21.6 g NaBr (47 mmol l−1) was dissolved in 4500 ml water and 45 g fibers were disintegrated in the solution at a pH of 10 (1 M NaOH). Then 13.4 g NaOCl (56.5 mmol l−1) was applied in the solution with five equal additions. The pH of the solution was kept at 10 by addition of NaOH solution (1 M) during the oxidation until the pH of the solution remained constant, which took ∼1.5 h. Then the oxidation reaction was stopped with an addition of 50 ml of ethanol. The pH of the solid dispersion was adjusted to 2.5 with 1 M HCl and the fibers were washed with pH 2.5 water three times by Buchner filtration followed by washing (three times) with deionizided water. This was intended to remove residual acid from the fibers. The resultant carboxylated fibers were then stored in a refrigerator. The carboxyl content of the fibers was investigated with the conductometric titration (shown below).Functionalization of carboxylated fibers
21.4 g (0.108 mol) of aminobenzophenone (addition levels equivalent to twice the carboxyl group content in fibers) was dissolved in a small volume of DMSO. Then 40 g of carboxylated fibres were diluted to a solids content of 15 g l−1 with pH 5 water. 20.8 g (0.108 mol) of EDC and 12.5 g NHS (0.108 mol) (both chemicals added as twice the carboxyl group content in fibers) were added to the dispersion, which was allowed to react with carboxylated fibers for 15 min, after which the dissolved aminobenzophenone was added. Then the dispersion was allowed to react overnight under darkness at room temperature. After the reaction, the benzophenone-functionalized fibers were thoroughly washed first with 50/50 mix of DMSO/water and then with deionized water. The grafting density was investigated with the conductometric titration and FTIR-measurements after microfluidization (shown below). Functionalized fibers were stored in a refrigerator.Microfluidization and film casting
The fibers with and without BP activation were microfluidized in a M110P fluidizer (Microfluidics corp., Newton, MA, USA). Fibers were diluted to 1.5% solids content in deionized water, the pH of the dispersion was adjusted to 9 with NaOH and the suspension was passed through the chamber pair (400 and 100 μm) once. The nanofibrillated cellulose gels were stored in a refrigerator. Nanofibrillated films were casted using pressurized filtration:3 diluted suspensions were filtered with a membrane under 2 bar pressure after which wet film were rolled five times with a smooth metal rolling pin for further consolidating the structure, and dried between clean blotting boards (blotting paper) under load and heat (100 °C) for 2 h. The effect of BP modification was also tested using a set of films prepared from mixtures of non-treated and BP-treated CNF suspensions. These films were prepared via simple solvent casing.UV activation of the films
Dry CNF films with and without BP were exposed to UV with 16 J cm−2 activation energy, using Uvitec CL 508L UV chamber (Cambridge, UK, UV wavelength 365 nm).Carboxyl content of TEMPO-oxidized fibers
The carboxyl content of TEMPO-oxidized fibers before and after conjugation of aminobenzophenone was determined by conductometric titration by following standard SCAN-CM 65:02. The determination was carried out with 0.25 g (dry pulp) fiber samples with 0.1 M NaOH with 0.025 ml injections with 30 s intervals. The amount of weak acid (carboxyl) groups was calculated as described in the standard method (SCAN-CM 65:02).ATR-FTIR and XPS analysis of dry CNF films
The chemistry of BP-TOCNF films was studied by using ATR-FTIR and XPS. The conjugation of aminobenzophenone onto TOCNF was evaluated with a Nicolet 750 Magna device as an attenuated total reflection (ATR). At least three areas in each sample were analysed. All spectra were obtained from 32 scans with a resolution of 4 cm−1 by using absorbance mode from 400 to 4000 cm−1.The surface composition of self-supporting CNF films was analysed by a Kratos Analytical AXIS Ultra X-ray photoelectron spectrometer with a monochromatic Al Kα X-ray source at 100 W and a neutralizer. The XPS experiments were performed with dry TOCNF films (pre-evacuated overnight). At least three spots of each sample were analyzed. Spectra were collected at an electron take-off angle of 90° from sample areas less than 1 mm in diameter. Elemental surface compositions were determined from low-resolution measurements (160 eV pass energy and 1 eV step) while the surface chemistry was probed with high resolution measurements (20 eV pass energy and 0.1 eV step). The carbon C 1s high-resolution spectra were curve fitted using parameters defined for cellulosic materials.19 All binding energies were referenced to the aliphatic carbon component of the C 1s signal at 285.0 eV. The UHV conditions remained satisfactory during the XPS experiments as per a reference used in situ (100% cellulose ash-free filter paper) that was measured along with each sample batch.
Water contact angle measurements
The changes in the hydrophobicity of TOCNF films with and without BP grafting was analysed with a contact angle goniometer CAM 200 (KSV instruments Ltd, Helsinki, Finland). Measurements were performed at room temperature with water as a probe liquid. A droplet volume of 6.5 μl and a recording time of 60 s were used to measure the time dependency of the contact angle. Water contact angles were measured on three locations on each sample.Surface morphology measurements with AFM
The surface topography of the materials with/without BP was analysed with a Nanoscope IIIa Multimode scanning probe microscope (Digital Instruments, Inc., Santa Barbara, CA). In this case, ultra-thin films were prepared by spin-coating diluted drops of neat and BP-grafted TOCNF onto PEI-coated gold wafers using a spinning rate of 3000 rpm for 1 min. Then the wafers carrying the samples were dried in the oven (80 °C for 10 min). Imaging was performed by using silicon cantilevers (Ultrasharp μmasch, Tallinn, Estonia). At least three different locations on each sample were scanned with image sizes of 5 × 5 μm2 and 1 × 1 μm2. No image processing was carried out except image flattening.Strength of TOCNF films
Tensile strength of TOCNF films with and without benzophenone were carried out with a MTS400/M vertical tensile tester (MTS system corporation, Eden Prairie, USA) with a 200 N load cell. The measurements were performed with 50 × 15 mm2 test strips, the 40 mm grip distance, and the testing velocity of 0.5 mm min−1 in a standard climate (50% RH, 23 °C). The thicknesses of TOCNF films were measured with a Lorentz wetter paper thickness meter. The average thickness values (average from three measurements of each sample) of TOCNF films were utilized to normalize the tensile forces when the tensile strength was calculated. The tensile strength was normalized with the mass density of the films in order to take into account the density variations of casted TOCNF films.The effect of BF content on material properties was evaluated using solvent cast films prepared from mixtures of neat and BP treated NCF. Mixed TOCNF/BP-TOCNF films with and without UV-activation were placed in water for 2 days and then the films were analysed visually while wet.
Wet strength under high energy sonication after 24 h exposure to water
The effect of BP treatment on the wet strength of TOCNF films was analysed with a tip sonicator (10% amplitude, 5 min time). TOCNF films with and without BP functionalization as well as with and without UV exposure (365 nm wavelength for 16 J) were kept in MilliQ water for one day, after which they were tip-sonicated with 10% amplitude and 2 min time.Results and discussion
In this work we investigated a method to conjugate the BP hydrophobic functionality onto highly hydrophilic TOCNF by using a sequential fiber pre-functionalization followed by mechanical disintegration. During TEMPO-oxidation, most of the residual hemicelluloses were removed.8 Moreover, it has been shown that TEMPO-oxidation reactants penetrate evenly in the cell wall of wood fibers, leading to uniform carboxyl group distribution throughout their structure.20 The carboxyl content of the TEMPO-oxidized fibres were 1.36 meq. g−1, comparable to values reported in the literature.21Benzophenone was grafted onto the carboxylated fibers via EDC/NHS activation from DMSO/water mixture. Since benzophenone is not water soluble, the reaction was carried out in water with the addition of a small amount of DMSO in which the BP was dissolved prior to the reaction. Note, DMSO has also been reported to swell wood fibers, possibly increasing the accessibility of the carboxylic groups on the cellulosic fibers to which the BP was linked through an amidation reaction.1 During the amidation reaction the fibers turned slightly yellow (Fig. 1a and b). After reaction, the BP-modified wood-fibers (Fig. 1c) were disintegrated with a fluidizer, and then the disintegrated BP-TOCNF was casted into the films by using pressurized filtration as described.3
 | ||
| Fig. 1 Photo images of (a) TEMPO-oxidized fibers, (b) benzophenone-functionalized fibers, and disintegrated BP-TOCNF (c). An AFM height image of BP-TOCNF is also included (the z-limit of the image is 14 nm) (d). | ||
The AFM height image of BP-TOCNF shows a typical fibrillar structure (Fig. 1d), which is rather similar to that of unmodified TOCNF (Fig. S1†).21 The BP-TOCNF were approximately 3–4 nm thick, as determined by the Nanoscope analysis software. Interestingly BP-TOCNF fibrils were slightly straighter than unmodified TOCNF fibrils. This could be explained if BP grafting altering TOCNF hydrophobicity which would make them more rigid when being in contact with water.
Carboxyl contents of TEMPO-oxidized fibers before and after conjugation of BP are shown in Fig. 2a. Due to the EDC/NHS coupling of the aminobenzophenone to the surface carboxyl groups the amount of charges decreased by ca. 70% during the reaction, indicating large degree of conversion of BP-linkage. However, significant amount of free carboxyl groups remained on the fiber surface. These would be beneficial in the following disintegration step, as they would improve colloidal stability and swelling.8 The amide bonds formed during the conjugation reaction were further investigated with ATR-FTIR, see Fig. 2b. Typical peaks for amide bonds were observed: 1645 cm−1 (amide I band, CO stretching) and 1549 cm−1 (amide II band, combined N–H deformation and C–N stretching), further confirming the linking of benzophenone. Moreover, the characteristic CO peak (1603 cm−1) of sodium carboxylate22 decreased remarkably when BP was grafted. We did not consider aldehydes produced during the alkaline TEMPO-oxidation that can raise the conjugation level of BP due to the nucleophile addition reaction between aldehydes and amines.23
 | ||
| Fig. 2 Carboxyl content of TEMPO-oxidized fibers before and after conjugation of BP by conductometric titration (a). FTIR absorbance spectra of TOCNF and BP-TOCNF (b). | ||
XPS was utilized to investigate the surface compositions of the TONCF films with and without BP, see Fig. 3. TEMPO oxidation showed up as marked increase in elemental sodium and in aliphatic/aromatic carbon C 1s component at 285 eV even though carboxylic groups cannot be reliably detected in cellulose derivatives by XPS.24,25 In the film with BP nitrogen (N 1s, 2 at%) was also detected, while BP treated samples otherwise exhibited characteristics typical to cellulose.
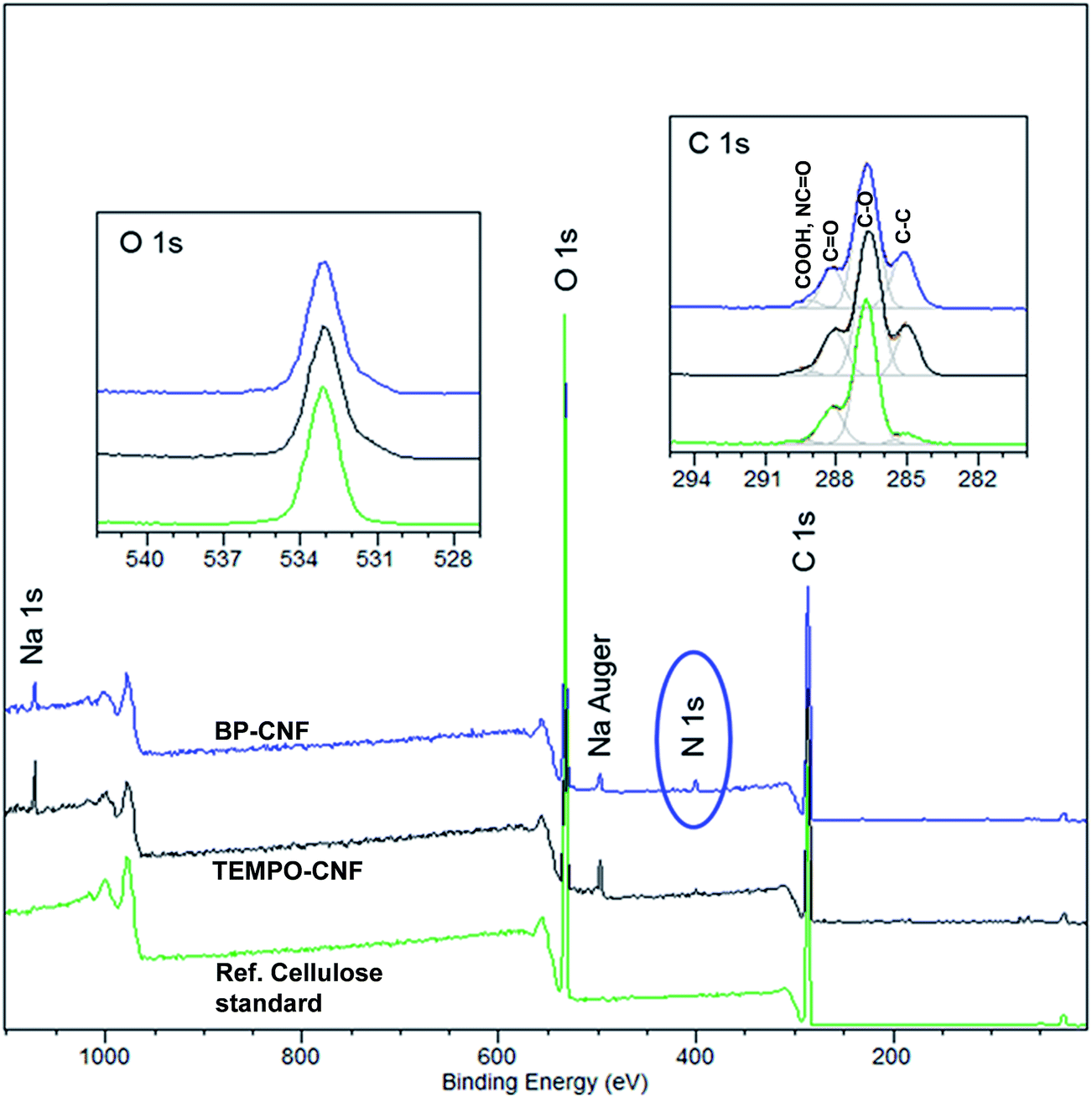 | ||
| Fig. 3 XPS spectra of TOCNF with and without benzophenone substituents. Included are also the respective spectra of a cellulose standard. | ||
Water contact angle (WCA) measurements were carried out on TOCNF films with and without grafted BP (Fig. 4). As expected, the contact angle of BP-TOCNF was remarkably higher than that of TOCNF (WCA after 30 s for TOCNF and BP-TOCNF were approximately 15 and 50 degrees, respectively). In the literature, CNF films before and after TEMPO-oxidation have been reported to be 10–35 and 40 degrees, respectively.3,26,27 The observed static CA for BP-TOCNF is, nevertheless, lower than that of hydrophobic octadecylamine substituents that raised the CA of TOCNF to 108 degrees.26 So, the FTIR, XPS, and WCA observations together yield evidence that BP was grafted onto the carboxylated fibrils.
 | ||
| Fig. 4 Water contact angles as a function of time of TOCNF (bottom curve) and BP-TOCNF (upper profile). | ||
The physical properties of pressure filtered TOCNF films with and without BP substituents are summarized in Table 1. The observed density for the non-modified TOCNF (1.005 g cm−3) was lower than the reported value for films made from mechanical disintegrated CNF, 1.080–1.200 g cm−3 and TEMPO-oxidized CNF, 1.450 g cm−3.28,29 This is probably due to the film manufacturing method: in this study films were prepared with the pressure filtration and while in the above mentioned studies solvent casting was typically utilised, and the latter leads to higher CNF film densities. However, the density of BP-TOCNF films was still much lower (only 0.664 g cm−3), and in addition to this, the BP grafted TOCNF films were much thicker (more than 50%) than the unmodified TOCNF films. Both observations suggest that hydrophobicity induced by BP grafting made the nanofibrillated fibres more rigid in water.
| Density (g cm−3) | Thickness (μm) | Specific tensile strength (MPa cm3 g−1) | Elongation (%) | |
|---|---|---|---|---|
| TEMPO-CNF | 1.0 ± 0.38 | 54 ± 2 | 109 ± 10 | 3.3 ± 0.6 |
| BP-CNF without UV | 0.66 ± 0.32 | 84 ± 4 | 114 ± 23 | 2.7 ± 1.2 |
| BP-CNF with UV | 0.70 ± 0.44 | 80 ± 5 | 138 ± 10 | 2.0 ± 0.5 |
The density variations were considered in the tensile strength measurements by normalizing the tensile strength by density (specific tensile strength). The strength values for the TOCNF observed are in the same range as reported in literature, 80–230 MPa for unmodified CNF,3,30 although direct comparison is difficult, due to variation in the experimental setup. Without UV-activation, no significant strengthening or changes in strain of the BP-TOCNF film were observed. However, after the UV-activation (UV-curing) the tensile strength of the BP-TOCNF films increased from 114 to 138 MPa cm3 g−1 while the elongation decreased slightly (from 2.7 to 2.0%). This observation strongly suggests the UV light catalysed radical crosslinking between cellulose nanofibrils and BP substituents.15
Preliminary tests on the effect of the UV-induced crosslinking on the BP-TOCNF film swelling in water was carried out, using films with different amounts of BP. The TOCNF/BP-TOCNF films with and without UV-activation were placed in water for two days and then the films were analysed visually while still wet (Fig. S2, ESI†). The thicknesses of the films without UV activation decreased slightly as a function of BP-TOCNF added (the thickness values were not measured since the films with low BP-content were too soft and brittle for accurate thickness measurements). This was due to higher hydrophobicity induced by BP. The effect of UV-induced crosslinking in the swelling of mixed films was remarkable: all films with added BP-TOCNF stayed thin compared to those without UV-activation. Furthermore, the thickness of a film made from 100% BP-TOCNF did not change during swelling in water, further supporting the hypothesis that UV-light induced covalent bond formation in BP-TOCNF films. In this study were did not consider the effect of wet pressing while the BP-TOCNF films were UV-activated. This could lead to a better crosslinking efficiency since fibrils are expected to be in closer proximity at the same time that radical-induced coupling reactions occur.
The effect of BP on CNF film stability in water was also investigated, using sonification tests. The sonication was carried out in the absence of mechanical contact with the tip, see images in Fig. S3.† As expected, films of TOCNF and BP-TOCNF without and with UV-activation disintegrated fully after immersion in water for 24 h and followed by 2 min sonication. Only the UV-activated BP-TOCNF film remained intact. Both, films of TOCNF and BP-TOCNF without UV-activation became dispersed within the first seconds of sonication. The observation that UV-induces BP-TOCNF crosslinking in the film correlated with the mechanical energy supplied to the film, which is roughly equal with results from BP-functionalized CNC.15 In this study BP was linked onto CNC through hydroxyl groups with 4-benzoylphenyl (6-isocyanatohexyl) carbamate (Bp-NCO), and no charged groups were introduced onto the CNC prior to the grafting of the BP groups. Further, in this latter study, the density of grafted hydroxyl groups was 157 μmol g−1, which is six fold lower compared to 957 μmol g−1, which we achieved in the presence of installed carboxyl groups. On the other hand, the large density of BP grafted on CNF reduces the hydrophilicity of the material (note the recorded contact angles). By adjusting the grafting conditions, it is possible to tune the interactions with water as required in different applications.
Conclusions
We have developed a strategy to graft benzophenone (BP) onto highly hydrophilic, TEMPO-oxidized cellulose nanofibrils. The grafting was achieved in spite of the high gel forming tendency and complex rheological behaviour of CNF dispersed in water. The developed method may be easily adapted for further grafting of other functional groups onto CNF fibrils, a task that will be followed in the future. The UV-induced crosslinking of BP-TOCNF made films strong even in humid or wet conditions. The UV-crosslinkable CNF is expected to be useful in applications where increased wet strength and resistance against light-induced aging are required.Acknowledgements
This work carried out under the sponsorship of TEKES-funded Design Driven World of Cellulose (DWOC) project in cooperation between Aalto University School of Chemical Technology and VTT, the Technical Research Centre of Finland. This work made use of Aalto University Bioeconomy XPS Facilities.References
- D. Klemm, Comprehensive cellulose chemistry. Volume 1, Fundamentals and analytical methods, Wiley-VCH, Weinheim, 1998 Search PubMed.
- M. Jorfi and E. J. Foster, J. Appl. Polym. Sci., 2015, 132, 41719 CrossRef.
- M. Österberg, J. Vartiainen, J. Lucenius, U. Hippi, J. Seppälä, R. Serimaa and J. Laine, ACS Appl. Mater. Interfaces, 2013, 5, 4640–4647 Search PubMed.
- M. Paakko, J. Vapaavuori, R. Silvennoinen, H. Kosonen, M. Ankerfors, T. Lindstrom, L. A. Berglund and O. Ikkala, Soft Matter, 2008, 4, 2492–2499 RSC.
- A. Walther, J. V. I. Timonen, I. Díez, A. Laukkanen and O. Ikkala, Adv. Mater., 2011, 23, 2924–2928 CrossRef CAS PubMed.
- S. Iwamoto, A. Isogai and T. Iwata, Biomacromolecules, 2011, 12, 831–836 CrossRef CAS PubMed.
- M. A. Hubbe and O. J. Rojas, BioResources, 2008, 3, 1419 Search PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- S. Spoljaric, A. Salminen, N. Luong and J. Seppälä, Cellulose, 2013, 20, 2991–3005 CrossRef CAS.
- T. Kurihara and A. Isogai, Cellulose, 2014, 21, 291–299 CrossRef CAS.
- G. Dorman and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef CAS PubMed.
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- M. Pérez-Perrino, R. Navarro, O. Prucker and J. Rühe, Macromolecules, 2014, 47, 2695–2702 CrossRef.
- C. Walling and M. J. Gibian, J. Am. Chem. Soc., 1965, 87, 3361–3364 CrossRef CAS.
- M. V Biyani, M. Jorfi, C. Weder and E. J. Foster, Polym. Chem., 2014, 5, 5716 RSC.
- A. Böhm, M. Gattermayer, C. Trieb, S. Schabel, D. Fiedler, F. Miletzky and M. Biesalski, Cellulose, 2013, 20, 467–483 CrossRef.
- M. Jocher, M. Gattermayer, H.-J. Kleebe, S. Kleemann and M. Biesalski, Cellulose, 2015, 22, 581–591 CrossRef CAS.
- A. Isogai and Y. Kato, Cellulose, 1998, 5, 153–164 CrossRef CAS.
- L.-S. Johansson and J. M. Campbell, Surf. Interface Anal., 2004, 36, 1018–1022 CrossRef CAS.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- H. Fukuzumi, T. Saito, Y. Okita and A. Isogai, Polym. Degrad. Stab., 2010, 95, 1502–1508 CrossRef CAS.
- G. T. Hermanson, Bioconjugate techniques, Academic Press, San Diego (CA), 2008, vol. 2 Search PubMed.
- J.-L. DiFlavio, R. Pelton, M. Leduc, S. Champ, M. Essig and T. Frechen, Cellulose, 2007, 14, 257–268 CrossRef CAS.
- J. Johansson, L.-S. Campbell, J. Orelma, H. Shchukarev and A. Laine, in Abstracts of 15th ECASIA 13.-18, 2013, p. 12 Search PubMed.
- R. Johnson, A. Zink-Sharp and W. Glasser, Cellulose, 2011, 18, 1599–1609 CrossRef CAS.
- H. Orelma, I. Filpponen, L.-S. Johansson, M. Österberg, O. Rojas and J. Laine, Biointerphases, 2012, 7, 1–12 CrossRef PubMed.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- J. Lucenius, K. Parikka and M. Österberg, React. Funct. Polym., 2014, 85, 167–174 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15015b |
| ‡ Present address: VTT, Technical Research Centre of Finland, Biologinkuja 7, P.O. Box 1000, FIN-02044 VTT, Finland. |
| This journal is © The Royal Society of Chemistry 2016 |