DOI:
10.1039/C6RA14915D
(Paper)
RSC Adv., 2016,
6, 91508-91516
BiOX/BiOY (X, Y = F, Cl, Br, I) superlattices for visible light photocatalysis applications
Received
8th June 2016
, Accepted 11th September 2016
First published on 14th September 2016
Abstract
The BiOX/BiOY (X, Y = F, Cl, Br, I, X ≠ Y) systems have been investigated as possible visible light photocatalysts in contrast with the BiOX (X = F, Cl, Br, I) systems by using hybrid density functional calculations. All the BiOX/BiOY systems have indirect bandgaps, and all the bandgaps of BiOX/BiOY systems we considered are between the bandgaps of BiOX and BiOY systems. The calculated bandgaps for BiOF/BiOCl, BiOF/BiOBr, BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI are respectively 3.86, 3.41, 2.74, 2.99, 2.30, and 2.23 eV. The maximum absorption wavelength increases in the order of BiOF, BiOF/BiOCl, BiOCl, BiOF/BiOBr, BiOCl/BiOBr, BiOBr, BiOF/BiOI, BiOCl/BiOI, BiOBr/BiOI, and BiOI. The conduction band edges for all the BiOX/BiOY systems originate from Bi 6p states, but the valance band edges are contributed by different electronic states. Besides, the relative positions of X p states and Y p states for BiOX/BiOY systems are different, which should be attributed to the different p orbital energies of X and Y atoms. Due to the conduction band maximum is lower than the hydrogen reduction potential, all the BiOX and BiOX/BiOY systems are thermodynamically unfavorable for hydrogen production. Meanwhile, owing to the suitable bandgaps and band edge positions, the BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI superlattices are possible visible light photocatalysts for degradation of organic pollutants.
1 Introduction
Using semiconductors for hydrogen storage, photodegradation of environment pollutants and photocatalytic decomposition of water into hydrogen has been an attractive way to solve the energy exhaustion and environment pollution issues.1–11 However, finding a suitable photocatalyst, which can efficiently make use of visible light for photocatalytic water splitting into hydrogen and degradation of pollutants, is still challenging. Doping with foreign elements including acceptor doping, donor doping, acceptor–donor codoping, and double-hole-mediated coupling codoping12–19 is one of the major strategies to modify the conventional wide bandgap semiconductors so as to extend the absorption range up to the visible light. In addition, adopting the methods such as noble metal loading,20–22 heterojunction constructing,23,24 and dye sensitizing25,26 generally make the semiconductors achieve better visible light response and efficient separation of the electron–hole pairs. Recently, many efforts have been devoted to develop the novel photocatalysts such as sulfides, nitrides, oxysulfides, oxynitrides, and oxyhalides etc. Among them, the bismuth oxyhalides (BiOX, X = F, Cl, Br, I) have drawn extensive attention in the area of heavy metal ions remove, sterilization, and photocatalytic degradation of organic pollutants.27–35 BiOX crystal belongs to the PbFCl-type tetragonal structure of space group P4/nmm (no. 129), and its structure is stacked by [–X–Bi–O–O–Bi–X–] layers, which are formed by the layer of [Bi2O2]2+ slabs interleaved by double slabs of halogen ions [X]−, by weak nonbonding van der Waals interaction through the halogen ions along the axis.36 The positive [Bi2O2]2+ and negative [X]− layers can induce an internal electric field along the c axis, and the formed internal electric field promoting the separation efficiency of photo-induced electron–hole pairs will be beneficial for the photocatalytic activity of BiOX. The bandgap of BiOCl has been evaluated to be 3.44,37 3.46,38 or 3.51 eV (ref. 39) according to the measured absorption spectra, while the experimental bandgaps for BiOBr and BiOI are respectively 2.91 (ref. 40) and 1.92 eV.39,40 On the basis of the research results by density functional theory, BiOF is a direct bandgap semiconductor, while the BiOCl, BiOBr, BiOI have indirect bandgaps.41–44 In addition, nanoparticles of BiOCl, BiOBr, and BiOI have been prepared in reverse microemulsions,45 and nano-sized BiOI has a bandgap up to 2.96 eV, which is quite different from the case of its bulk material.
Recent years many BiOX based heterojunctions and nanocomposites have been fabricated and studied in order to enhance the photocatalytic efficiency for solar energy conversion. For instance, the BiOCl/SnO2 heterojunction photocatalysts have been designed and prepared through a two-step solution route with SnO2 nanoparticles dispersed on the BiOCl(001) plane,46 the nanocomposites exhibit higher photocatalytic activity as compared to the individual BiOCl or SnO2 in the degradation of rhodamine B under visible light irradiation. The reason for the enhanced photocatalysis for BiOCl/SnO2 composites should be attributed to the fact that the formed defects states and p–n heterojunctions decrease the bandgap and promote the separation efficiency of photoinduced electron–hole pairs. A novel Bi2Si3/BiOCl47 hybrid architectures which are sensitive to visible light have been synthesized through a controlled anion exchange approach. Heterojunction BiOBr/BiVO4 photocatalysts have been prepared through acid etching coupled with a hydrothermal process,48 the BiOBr/BiVO4 composites show a higher visible light photocatalytic performance in degradation of MB as compared to the single BiOV4 and BiOBr. The visible light responsive BiOBr/ZnFe2O4 (ref. 49) heterojunction photocatalysts have been designed and prepared by an ultrasound-assisted precipitation–deposition method, the composites exhibit exceptional photocatalytic activity in the degradation of rhodamine B under visible light irradiation. The study by Sun et al.50 has shown that BiOF/Bi2O3 nanostructures have been fabricated by hydrolysis method, and the prepared BiOF/Bi2O3 exhibits superior performance in visible light degradation of rhodamine B. In addition, some other BiOX based composites, such as CoFe2O4/BiOCl, CoFe2O4/BiOBr, CoFe2O4/BiOI,51 SiO2/BiOCl, SiO2/BiOBr, SiO2/BiOI,52 WO3/BiOCl,53 RGO/BiOCl (reduced graphene oxide/BiOCl),54 TiO2/BiOBr,55 BiOCl/Bi4Ti3O12,56 MoS2/BiOBr,57 BiOBr/ZnO58 etc., have been found to extend the light absorption to the visible light region and to display high visible light photocatalytic activity. Besides, heterojunctions can be formed among BiOCl, BiOBr, and BiOI. The BiOCl/BiOBr composites with different Cl-to-Bl molar rations have been synthesized using a simple microwave-assisted coprecipitation method,59 and the visible light photocatalytic activity in degradation of rhodamine B of such composites are significantly enhanced due to the strong absorption of the visible light and the reduced recombination rate of electron–hole. BiOI(001)/BiOCl(001) and BiOI(001)/BiOCl(010) heterojunctions have been designed and synthesized with a crystal facet engineering method,60 and both of these BiOI/BiOCl heterojunctions have higher visible light photocatalytic activity than the single BiOI or BiOCl due to the formed heterojunctions could obviously reduce the recombination of electron–hole pairs and promote the separation of electron–hole pairs. BiOI/BiOBr61 films have been prepared by a facile solvothermal method, and the photocatalytic activity in degradation of methyl orange of BiOI/BiOBr has been significantly enhanced as compared to pure BiOBr film in the decomposition of methyl orange. Some other studies62–67 also indicate that BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI heterojunctions have high visible light photocatalytic performance in degradation of rhodamine B and methyl orange.
There are a large number of experiment reports about the photocatalytic capability of BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI heterojunctions and composites, so it will be meaningful to gain into the physical mechanism of the high visible light photocatalytic ability of them. Because F has the highest electronegativity and can significantly affect the electron distribution by strongly adsorb or trap electrons, F could effectively improve the photocatalytic ability.68 Though there is almost no report about the heterojunction composites of BiOF/BiOCl, BiOF/BiOBr, and BiOF/BiOI, it will be surprising to find whether the BiOF/BiOCl, BiOF/BiOBr, and BiOF/BiOI systems have the high visible light photocatalytic ability. In this paper, we mainly calculate the structural, electronic, and optical properties of BiOX/BiOY (X, Y = F, Cl, Br, I; X ≠ Y) heterostructures as compared with the results of the individual BiOX systems. So as to facilitate the calculation and processing, herein we build the (001) BiOX1 unitcell/BiOY1 unitcell heterostructure models, i.e., BiOX/BiOY superlattices. The structure of this work is organized as follows. Section 2 gives the details of computational method we adopted in this work, while Section 3 displays the results and discussion about the structural, electronic, and optical properties of BiOX and BiOX/BiOY systems, and finally we offer some concluding remarks based on the results in Section 4.
2 Computational details
BiOX crystal belongs to the PbFCl-type tetragonal structure of space group P4/nmm (no. 129),70 and the unit cell of unrelaxed BiOX containing 2 Bi, 2 O, 2 X atoms is illustrated in Fig. 1. The BiOX/BiOY superlattice system is constructed by substituting one monolayer of two (001) monolayers of BiOX by one BiOY(001) monolayer and the unrelaxed BiOX/BiOY superlattice model is displayed in Fig. 2. Projector augmented wave (PAW) method71 has been adopted to perform first-principles calculations as implemented in the Vienna Ab initio Simulation (VASP) Package.72,73 Generalized gradient approximation (GGA)74 with Perdew–Burke–Ernzerhof (PBE) functional75 for the exchange correlation contribution has been adopted for structural optimization and total energy calculation. The valence electronic configurations for Bi, O, F, Cl, Br, I are respectively 6s26p3, 2s22p4, 2s22p5, 3s23p5, 4s24p5, 5s25p5. Because the unique PbFCl-type layer structure of BiOX system with the tetragonal space group P4/nmm (no. 129) is stacked together with [–X–Bi–O–Bi–X–] layers by weak van-der-Waals interactions through X atoms along the c axis, van-der-Waals correction proposed by Grime (DFT-D3)76 is adopted for good description of long-range van-der-Waals interaction. The plane-wave cutoff of 500 eV, the tolerance for energy convergence of 1.0 × 10−6 eV, the convergence thresholds for the force on the atoms of 10−2 eV Å−1, the Monkhorst–Pack k-point77 of 16 × 16 × 8 for BiOX systems and the Monkhorst–Pack k-point of 16 × 16 × 4 for BiOX/BiOY systems have been sufficient for geometry optimization and total energy calculation. To accurately describe the band structures, density of states (DOS) and optical properties, the more time-consuming Heyd–Scuseria–Ernzerhof (HSE06)78,79 hybrid functional has been adopted. The electron–electron interaction energy for HSE functional consists of short-ranged (SR) and long-ranged (LR) parts:| | |
EHSEXC = χESRX(μ) + (1 − χ)EPBE,SRX(μ) + EPBE,LRX(μ) + EPBEC,
| (1) |
where μ is the screening parameter and χ is the mixing coefficient. Here the μ and χ are respectively determined as 0.20 Å−1 and 0.20 to obtain the bandgaps of BiOX systems close to the experimental values. In addition, k-point of 8 × 8 × 4 for BiOX systems and 8 × 8 × 2 for BiOX–BiOY systems have been found sufficient for the HSE06 calculations, and the absorption curves can be obtained from the imaginary part of the dielectric constant using Kramers–Kroning dispersion relation.80
 |
| | Fig. 1 Illustration of the unrelaxed BiOX (X = F, Cl, Br, I). | |
 |
| | Fig. 2 Illustration of the unrelaxed BiOX/BiOY (X, Y = F, Cl, Br, I) superlattices. | |
The CBM positions for the BiOX and BiOX/BiOY systems with respective to energy levels of water reduction potential of H+/H2 (0 eV versus normal hydrogen electrode, i.e., 0 eV vs. NHE) and the water oxidation potential of O2/H2O (1.23 eV) could be estimated according to the method developed by Butler and Ginley:81–83
| | |
ECBM(BiOX) = (χBi2χO2χX2)1/6 − 0.5Eg(BiOX) + EO,
| (2) |
| | |
EVBM(BiOX) = ECBM(BiOX) + Eg(BiOX),
| (3) |
| | |
ECBM(BiOX/BiOY) = (χBi4χO4χX2χY2)1/12 − 0.5Eg(BiOX/BiOY) + EO,
| (4) |
| | |
EVBM(BiOX/BiOY) = ECBM(BiOX/BiOY) + Eg(BiOX/BiOY),
| (5) |
where
ECBM(BiOX) and
ECBM(BiOX) are respectively the energy levels of CBM and VBM for BiOX systems,
ECBM(BiOX/BiOY) and
EVBM(BiOX/BiOY) respectively denote the energy levels of CBM and VBM for BiOX/BiOY systems.
χO,
χBi,
χX,
χY denote the absolute electronegativity of O, Bi, X, and Y atoms, respectively. For BiOCl, BiOBr, BiOI systems,
Eg(BiOX) are respectively the experimental bandgaps of 3.51, 2.91, and 1.92 eV. For the BiOF, BiOF/BiOCl, BiOF/BiOBr, BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI systems, the values of
Eg(BiOX/BiOY) are obtained from the HSE06 calculations with the mixing coefficient of 0.20.
EO, which denotes the scale factor relating the reference electrode redox level to the absolute vacuum scale, is −4.5 eV for normal hydrogen electrode. Besides,
χO,
χBi,
χF,
χCl,
χBr,
χI are respectively 7.54, 4.69, 10.41, 8.30, 7.59, and 6.76 eV.
84
3 Results and discussion
Our calculated lattice parameters, the experimental lattice parameters, and some other relaxed lattice parameters for BiOX systems are summarized in Table 1. Using the above-mentioned computational method with considering the van der Waals interaction, the optimized lattice parameters for BiOF, BiOCl, BiOBr, and BiOI are well consistent with the experimental measurements40,69 and the other theoretical calculated results.41–43 For BiOX/BiOY superlattice systems, the space group and optimized parameters are listed in Table 2. The lattice constant of a (and b) for BiOX/BiOY is between lattice constants of a for BiOX and BiOY, and the lattice of c for BiOX/BiOY is almost equal to the sum of the c for BiOX and c for BiOY.
Table 1 Calculated space groups and lattice parameters for BiOX and BiOX/BiOY (X, Y = F, Cl, Br, I) systems
| Systems |
BiOF |
BiOCl |
BiOBr |
BiOI |
| Space group |
P4/nmm(129) |
P4/nmm(129) |
P4/nmm(129) |
P4/nmm(129) |
| a = b (Å) |
3.765 |
3.902 |
3.932 |
3.932 |
| c (Å) |
6.163 |
7.331 |
8.101 |
8.101 |
| Ref. 69 (Exp.) |
— |
3.89 |
— |
— |
| — |
7.38 |
— |
— |
| Ref. 40 (Exp.) |
— |
— |
3.9231 |
3.9967 |
| — |
— |
8.0945 |
9.1533 |
| Ref. 41 (Cal.) |
— |
3.757 |
3.794 |
7.961 |
| — |
7.198 |
7.961 |
8.980 |
| Ref. 42 (Cal.) |
3.7386 |
3.8743 |
3.8996 |
3.9738 |
| 6.1714 |
7.3997 |
8.4570 |
9.3722 |
| Ref. 43 (Cal.) |
3.748 |
3.891 |
3.916 |
3.985 |
| 6.224 |
7.369 |
8.077 |
9.129 |
Table 2 Calculated space groups, lattice parameters and bandgaps for BiOX and BiOX/BiOY (X, Y = F, Cl, Br, I) systems
| Systems |
Space group |
a = b/c (Å) |
Bandgap |
| BiOF/BiOCl |
P4mm(99) |
3.847/13.506 |
3.86 |
| BiOF/BiOBr |
P4mm(99) |
3.869/14.306 |
3.41 |
| BiOF/BiOI |
P4mm(99) |
3.917/15.220 |
2.74 |
| BiOCl/BiOBr |
P4mm(99) |
3.917/15.452 |
2.99 |
| BiOCl/BiOI |
P4mm(99) |
3.952/16.438 |
2.30 |
| BiOBr/BiOI |
P4mm(99) |
3.965/17.215 |
2.23 |
The bandgaps for BiOX calculated by hybrid density functional of HSE06 with different mixing coefficients and PBE functional, the bandgaps of experimental results,36,85 and other theoretical results41–44 are listed in Table 3. The results indicate that the bandgaps for BiOCl and BiOBr obtained by using HSE06 functional with χ = 0.20 are much closer to the experimental values,37–40 while the bandgap for BiOI obtained by using χ = 0.15 is much closer to the experimental values.39,40 Here we use HSE06 functional with χ = 0.20 to calculate BiOX–BiOY systems to ensure most of the bandgaps for BiOX systems much closer to the experimental values. The band structures, DOS and PDOS for BiOX systems are illustrated in Fig. 3 and 4. It can be clearly seen that both the valence band maximum (VBM) and conduction band minimum (CBM) for BiOF locate at the same k-point of Z, which indicates BiOF is a direct bandgap semiconductor. The calculated bandgap for BiOF of 4.18 eV (Table 3) is a little larger than the calculated value of 3.987 eV given by Zhao et al.42 by using GGA+U method and obviously larger than the other theoretical results43,44 obtained by using GGA-PBE functional, as GGA-PBE method seriously underestimates the bandgap value. The DOS and PDOS of BiOF is displayed in Fig. 4(a), the VBM is contributed by occupied Bi 6s, Bi 6p, O 2p, and F 2p states, while the CBM is mainly comprised of unoccupied Bi 6p states.
Table 3 The calculated and experimental bandgaps for BiOX and BiOX/BiOY (X, Y = F, Cl, Br, I) systems
| Systems |
BiOF |
BiOCl |
BiOBr |
BiOI |
| PBE |
3.12 |
2.60 |
2.12 |
1.61 |
| HSE (χ = 0.25) |
4.43 |
3.66 |
3.08 |
2.37 |
| HSE (χ = 0.20) |
4.18 |
3.45 |
2.89 |
2.21 |
| HSE (χ = 0.15) |
3.93 |
3.24 |
2.70 |
2.05 |
| Ref. 37 (Exp.) |
— |
3.44 |
— |
— |
| Ref. 38 (Exp.) |
— |
3.46 |
— |
— |
| Ref. 39 (Exp.) |
— |
3.51 |
— |
1.92 |
| Ref. 40 (Exp.) |
— |
— |
2.91 |
1.92 |
| Ref. 41 (Cal.) |
— |
2.50 |
2.10 |
1.59 |
| Ref. 42 (Cal.) |
3.987 |
3.504 |
2.865 |
1.906 |
| Ref. 43 (Cal.) |
3.22 |
2.80 |
2.36 |
1.75 |
| Ref. 44 (Cal.) |
3.41 |
2.69 |
2.21 |
1.62 |
 |
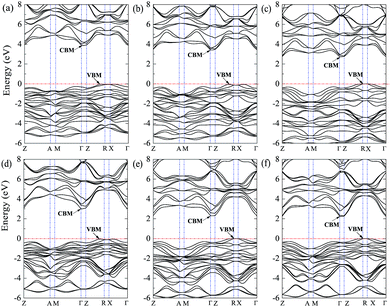
| | Fig. 3 Band structures of (a) BiOF, (b) BiOCl, (c) BiOBr, and (d) BiOI. The horizontal dashed lines represent the Fermi levels. | |
 |
| | Fig. 4 DOS and PDOS for (a) BiOF, (b) BiOCl, (c) BiOBr, and (d) BiOI systems. The vertical black dashed lines represent the Fermi levels. | |
The band structures for BiOCl, BiOBr, BiOI are displayed in Fig. 3(b)–(d), the VBM and CBM for these three crystals locate at different k-point positions, indicating BiOCl, BiOBr, BiOI are all indirect semiconductors. The calculated bandgap for BiOCl of 3.45 eV (Table 3) is in agreement with the experiment value37–39 and the previously calculated bandgap given by Zhao et al. by utilization of GGA+U method,42 while the bandgaps given by some other research groups41,43,44 are obviously smaller than the experiment measurements due to the underestimation of bandgap using the GGA-PBE method. Besides, the VBM and CBM are respectively located on the k-point line of Z → R and Z. The DOS and PDOS in Fig. 4(b) indicate that the VBM for BiOCl is composed of Bi 6s, Bi 6p, O 2p, and Cl 3p states, whereas the CBM is predominantly contributed by Bi 6p states. For the case of BiOBr, the calculated bandgap is 2.89 eV (Table 3) with VBM locating on the k-point line of Z → R and CBM locating on the k-point of Z. Our obtained bandgap is consistent with the experiment value40 and the calculated value given by Zhao et al.,42 but obviously larger than previously calculated bandgaps41,43,44 by using GGA-PBE method. The DOS and PDOS of BiOBr are illustrated in Fig. 4(c), the VBM is mainly composed of Bi 6s, Br 4p, O 2p states with a small amount of Br 4s, Bi 6p states. Seen from Fig. 3(d), the bandgap of BiOI is 2.21 eV (Table 3), and the k-point positions of VBM and CBM are the same as BiOCl and BiOBr in the primitive Brillouin zone. Our calculated bandgap is larger than the experimental value39,40 of 1.92 eV and the previously theoretical result of 1.906 eV by Zhao et al.42 Some other theoretical results41,43,44 by using GGA-PBE method are obviously smaller than the experimental value. It can be clearly seen from the DOS and PDOS of BiOI in Fig. 4(d) that the VBM is dominated by I 5p, O 2p, Bi 6s states with a small amount of I 5s, Bi 6p states, while the CBM is mainly composed of Bi 6p states.
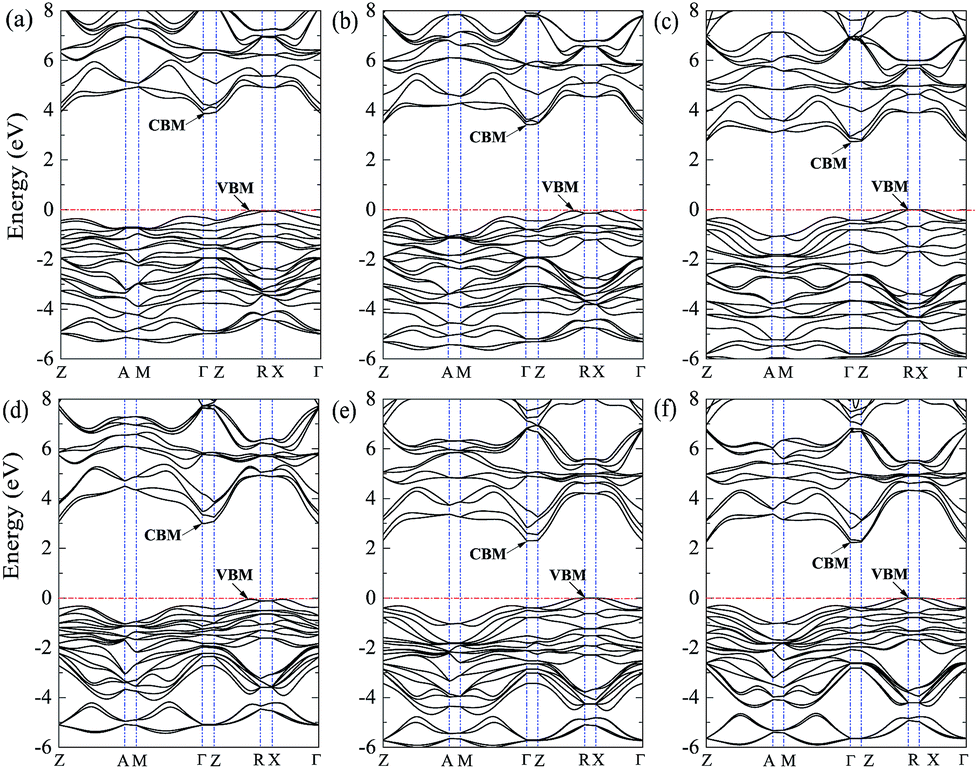
The band structures, DOS and PDOS for BiOX/BiOY systems are illustrated in Fig. 5 and 6. For the case of BiOF/BiOCl, the VBM is located on the k-point line of Z → R which is the same as the case of BiOCl, whereas the CBM is located on the k-point of Γ which is quite different from the case of BiOF and BiOCl whose CBM are located on the k-point of Z. The obtained bandgap of 3.86 eV (Table 2) for BiOF/BiOCl is between the bandgap values for BiOF and BiOCl. As displayed in Fig. 6(a), the CBM is mainly composed of Bi 6s, O 2p, Cl 3p states with a small amount of Bi 6p and F 2p states and the VBM mainly originates from the Bi 6p states. As shown in Fig. 5(b), the calculated VBM and CBM for BiOF/BiOBr are located at Γ and the k-point line of Z → R. The calculated bandgap for BiOF/BiOBr is 3.41 eV (Table 2), which is smaller than the bandgap of BiOF/BiOCl. Furthermore, the bandgap for BiOF/BiOBr is smaller than the bandgap for BiOF but larger than that of BiOBr. The DOS and PDOS for BiOF/BiOBr are displayed in Fig. 6(b), the VBM is predominantly comprised of Bi 6s, O 2p, Br 4p states, but there is almost no Bi 6p, F 2p states around the Fermi level, which is quite different from the case of BiOF/BiOCl. This could be attributed to the fact that the Br 4p orbital energy is much higher than the F 2p orbital energy. Besides, the CBM for BiOF/BiOBr is mainly composed of Bi 6p states. The calculated band structures, DOS, and PDOS for BIOF/BiOI are depicted in Fig. 5(c) and 6(c). It is found that the BiOF/BiOI is an indirect bandgap semiconductor with its CBM and VBM located at the different k-point positions. The bandgap for BiOF/BiOI of 2.74 eV (Table 2) is smaller than the bandgaps of BiOF/BiOCl and BiOF/BiOBr. Similarly, the bandgap is between the bandgaps for BiOF and BiOI. The VBM is mainly composed of I 5p with small amount of Bi 6s, O 2p states, and there is almost no F 2p states and Bi 6p states around the Fermi level. Similar to the case of BiOF/BiOCl and BiOF/BiOBr, the VBM of BiOF/BiOI is mainly comprised of Bi 6p states. It can be clearly seen that, the peaks of F 2p states in valance band are much lower than the Fermi level as compared with the cases of BiOF/BiOCl and BiOF/BiOI, which should be explained by the reason the difference between I 5p orbital and F 2p orbital is larger than that between Br 4p (Cl 3p) orbital and F 2p orbital.
 |
| | Fig. 5 Band structures of (a) BiOF/BiOCl, (b) BiOF/BiOBr, (c) BiOF/BiOI, (d) BiOCl/BiOBr, (e) BiOCl/BiOI, and (f) BiOBr/BiOI systems. The horizontal dashed lines represent the Fermi levels. | |
 |
| | Fig. 6 DOS and PDOS for (a) BiOF/BiOCl, (b) BiOF/BiOBr, (c) BiOF/BiOI, (d) BiOCl/BiOBr, (e) BiOCl/BiOI, and (f) BiOBr/BiOI systems. The vertical black dashed lines represent the Fermi levels. | |
As displayed in Fig. 5(d), the BiOCl/BiOBr is also an indirect semiconductor with the VBM and CBM locating on the different positions of k-point, and the bandgap is 2.99 eV (Table 2), which is larger than the bandgap of BiOBr but smaller than the bandgap of BiOCl. Zhang et al.59 have experimentally prepared BiOCl/BiOBr power composites with different Br/Cl molar rations, the bandgaps for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2.5, 1:5, 1:7.5, and 1:10 BiOCl/BiOBr composites are 3.26, 3.12, 2.94, 3.00, and 3.00 eV, respectively. Besides, another experimental study by Li et al.63 has prepared BiOCl/BiOBr microspheres with the bandgap of 2.88 eV. Our calculated bandgap of 2.99 eV for BiOCl/BiOBr superlattice is close to these experimental bandgaps for these BiOCl/BiOBr composites, which indicates that the BiOCl/BiOBr superlattice could be used to discuss the possible mechanism of the high photocatalytic activity of BiOCl/BiOBr composites in degradation of rhodamine B and methyl orange. The VBM of BiOCl/BiOBr superlattice is mainly contributed by Br 4p, Bi 6s, and O 2p states, and there is no appearance of Bi 6p and Cl 3p states around the Fermi level. This is mainly because the Br 4p orbital energy is higher than the Cl 3p orbital energy. The CBM is only composed of Bi 6p states. The band structures, DOS, and PDOS of BiOCl/BiOI are presented in Fig. 5(e) and 6(e), the BiOCl/BiOI has an indirect bandgap of 2.30 eV (Table 2), which is between the bandgaps of BiOCl and BiOI. The bandgap of 2.30 eV for BiOCl/BiOI superlattice is also close to the experimental bandgap of 1.98 eV for 20% BiOCl/BiOI composites and the experimental bandgap of 2.03 eV for 70% BiOCl/BiOI composites.64 The VBM is mainly contributed by I 5p states with small amount of O 2p and Bi 6s states, there is no appearance of Bi 6p and Cl 3p states below the Fermi level. This should be attributed to the lower Cl 3p orbital energy as compared to that of I 5p. The CBM is comprised of Bi 6p states. The band structures, DOS, and PDOS of BiOBr/BiOI are presented in Fig. 5(f) and 6(f), the BiOBr/BiOI has an indirect bandgap of 2.23 eV (Table 2), which is between the bandgaps of BiOBr and BiOI. The calculated bandgap of 2.23 eV is also located the range of experimental bandgaps for BiOBr/BiOI composites from 1.72 to 2.75 eV.66 The VBM is mainly contributed by I 5p states with small amount of O 2p and Bi 6s states, there is no appearance of Bi 6p and Br 4p states below the Fermi level. This may be due to the fact that the Br 4p orbital energy is lower than that of I 5p orbital energy. The CBM is comprised of Bi 6p states.
:1, 1:2.5, 1:5, 1:7.5, and 1:10 BiOCl/BiOBr composites are 3.26, 3.12, 2.94, 3.00, and 3.00 eV, respectively. Besides, another experimental study by Li et al.63 has prepared BiOCl/BiOBr microspheres with the bandgap of 2.88 eV. Our calculated bandgap of 2.99 eV for BiOCl/BiOBr superlattice is close to these experimental bandgaps for these BiOCl/BiOBr composites, which indicates that the BiOCl/BiOBr superlattice could be used to discuss the possible mechanism of the high photocatalytic activity of BiOCl/BiOBr composites in degradation of rhodamine B and methyl orange. The VBM of BiOCl/BiOBr superlattice is mainly contributed by Br 4p, Bi 6s, and O 2p states, and there is no appearance of Bi 6p and Cl 3p states around the Fermi level. This is mainly because the Br 4p orbital energy is higher than the Cl 3p orbital energy. The CBM is only composed of Bi 6p states. The band structures, DOS, and PDOS of BiOCl/BiOI are presented in Fig. 5(e) and 6(e), the BiOCl/BiOI has an indirect bandgap of 2.30 eV (Table 2), which is between the bandgaps of BiOCl and BiOI. The bandgap of 2.30 eV for BiOCl/BiOI superlattice is also close to the experimental bandgap of 1.98 eV for 20% BiOCl/BiOI composites and the experimental bandgap of 2.03 eV for 70% BiOCl/BiOI composites.64 The VBM is mainly contributed by I 5p states with small amount of O 2p and Bi 6s states, there is no appearance of Bi 6p and Cl 3p states below the Fermi level. This should be attributed to the lower Cl 3p orbital energy as compared to that of I 5p. The CBM is comprised of Bi 6p states. The band structures, DOS, and PDOS of BiOBr/BiOI are presented in Fig. 5(f) and 6(f), the BiOBr/BiOI has an indirect bandgap of 2.23 eV (Table 2), which is between the bandgaps of BiOBr and BiOI. The calculated bandgap of 2.23 eV is also located the range of experimental bandgaps for BiOBr/BiOI composites from 1.72 to 2.75 eV.66 The VBM is mainly contributed by I 5p states with small amount of O 2p and Bi 6s states, there is no appearance of Bi 6p and Br 4p states below the Fermi level. This may be due to the fact that the Br 4p orbital energy is lower than that of I 5p orbital energy. The CBM is comprised of Bi 6p states.
The calculated absorption spectra for BiOX and BiOX/BiOY systems are displayed in Fig. 7. Though the calculated maximum absorption wavelengths for BiOCl, BiOBr, BiOI by using HSE06 are obviously smaller than the experimental results,37–40 it does not affect the relative relationship of maximum absorption wavelengths for BiOX and BiOX/BiOY systems. For BiOF, BiOCl, BiOBr, and BiOI, the absorption edges shift towards the longer wavelength obviously with the X atomic numbers increasing. The maximum absorption wavelength for BiOX/BiOY system is between the maximum absorption wavelengths for BiOX and BiOY systems. The maximum absorption wavelength increases in the order of BiOF, BiOF/BiOCl, BiOCl, BiOF/BiOBr, BiOCl/BiOBr, BiOBr, BiOF/BiOI, BiOCl/BiOI, BiOBr/BiOI and BiOI. All the BiOX/BiOY systems have potential application as the visible photocatalytic materials.
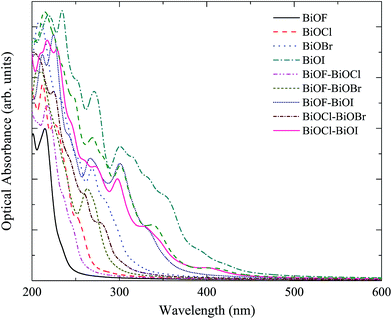 |
| | Fig. 7 The calculated optical absorption curves for BiOX and BiOX/BiOY (X, Y = F, Cl, Br, I) systems. | |
Not only the bandgaps but also the band edge positions are important to determine the photocatalytic activity of BiOX and BiOX/BiOY systems. The calculated VBM and CBM positions for BiOX and BiOX/BiOY systems with reference to the energy level of water reduction potential are illustrated in Fig. 8. As for all the BiOX and BiOX/BiOY systems, the CBM is lower than the hydrogen reduction potential, so it is thermodynamically unfavorable for hydrogen production. Almost no reports about BiOX/BiOY heterostructures used for photocatalytic water splitting to obtain hydrogen also demonstrates this. In addition, the CBM for the BiOX and BiOX/BiOY systems are not negative enough to reach the reduction potential of oxygen and to produce superoxide radicals (e− + O2 + H+ → HO2, −0.046 V vs. NHE).86 Therefore, the formation of superoxide radicals is irrelevant to the oxidation of BiOX and BiOX/BiOY systems. It is well known that the hydroxyl radicals (˙OH), as the oxidative species formed from the reaction between hole and H2O (h+ + H2O → ˙OH + H+, E°(˙OH/H2O) = +2.80 V vs. NHE),87–89 play significant roles in the photodegradation of organic pollutants, such as rhodamine B and methyl orange. Though the bandgap of BiOBr is obviously larger than that of BiOI, the photocatalytic ability of BiOBr in degradation of rhodamine B and methyl orange is higher as BiOBr has the suitable band edge positions. For BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, BiOBr/BiOI, their bandgaps are respectively 2.74, 2.99, 2.30, and 2.23 eV, so the visible light can be absorbed. They are suitable for photocatalytic degradation of organic pollutants, such as rhodamine B and methyl orange, as they have suitable band edge positions. The previous experiments59–67 also indicate that BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI exhibit high visible light photocatalytic activity in degradation of rhodamine B and methyl orange.
 |
| | Fig. 8 Schematic representation of the VBM and CBM positions for BiOX and BiOX/BiOY (X, Y = F, Cl, Br, I) systems. The energy level of water reduction potential of H+/H2 is set as energy zero, and the blue and green colors respectively denote VBM and CBM. | |
4 Conclusions
Hybrid density functional calculations have been performed to investigate the structural, electronic, and optical properties of BiOF/BiOCl, BiOF/BiOBr, BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI superlattice systems in contrast to the BiOF, BiOCl, BiOBr, and BiOI systems. The BiOX/BiOY (X, Y = F, Cl, Br, I, X ≠ Y) superlattice systems are constructed by substituting one monolayer of two (001) monolayers of BiOX by one BiOY(001) monolayer. Our calculated results indicate that all the BiOX/BiOY superlattice systems are indirect bandgap semiconductors, and the bandgaps of BiOX/BiOY systems are between bandgap values for BiOX and BiOY systems. The calculated bandgaps for BiOF/BiOCl, BiOF/BiOBr, BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI are respectively 3.86, 3.41, 2.74, 2.99, 2.30, and 2.23 eV. The conduction band edges for BiOX/BiOY systems all originate from Bi 6p states, but the valance band edges are composed by different electronic states. Besides, the relative positions of X p states and Y p states for BiOX/BiOY systems are different, which should be attributed to the different p orbital energies of X and Y atoms. The sequence of the maximum absorption wavelength is BiOF < BiOF/BiOCl < BiOCl < BiOF/BiOBr < BiOCl/BiOBr < BiOBr < BiOF/BiOI < BiOCl/BiOI < BiOBr/BiOI < BiOI. Due to the conduction band maximum is lower than the hydrogen reduction potential, all the BiOX and BiOX/BiOY systems are thermodynamically unfavorable for hydrogen production. Meanwhile, the suitable bandgaps and band edge positions make BiOF/BiOI, BiOCl/BiOBr, BiOCl/BiOI, and BiOBr/BiOI superlattices possible visible light photocatalysts for degradation of organic pollutants.
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 11175146 and 10904125, the Natural Science Foundation of Chongqing under Grant No. CSTC-2011BA6004 and CSTC-2008BB4253 and the Fundamental Research Funds for the Central Universities under Grant No. XDJK2012C038, XDJK2014D044 and XDJK2015C045.
References
- G. Wang, H. Yuan, A. Kuang, W. Hu, G. Zhang and H. Chen, Int. J. Hydrogen Energy, 2014, 39, 3780–3789 CrossRef CAS.
- Q. Wang, Q. Sun, P. Jena and Y. Kawazoe, ACS Nano, 2009, 3, 621–626 CrossRef CAS PubMed.
- S. J. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- T. Wang, Z. Luo, C. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7469–7484 RSC.
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50, 10768–10777 RSC.
- J. H. Pan, H. Dou, Z. Xiong, C. Xu, J. Ma and X. Zhao, J. Mater. Chem., 2010, 20, 4512–4528 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- C. Yu, W. Zhou, J. C. Yu, H. Liu and L. Wei, Chin. J. Catal., 2014, 35, 1609–1618 CrossRef CAS.
- C. Yu, W. Zhou, L. Hong, L. Yuan and D. D. Dionysiou, Chem. Eng. J., 2016, 287, 117–129 CrossRef CAS.
- C. L. Yu, J. C. Yu, H. B. He and W. Q. Zhou, Rare Met., 2016, 211–222 CrossRef CAS.
- Z. Zhao, X. He, J. Yi, C. Ma, Y. Cao and J. Qiu, RSC Adv., 2013, 3, 84–90 RSC.
- X. Wu, S. Yin, Q. Dong and T. Sato, Phys. Chem. Chem. Phys., 2013, 15, 20633–20640 RSC.
- B. Modak and S. K. Ghosh, Phys. Chem. Chem. Phys., 2015, 17, 15274–15283 RSC.
- G. Wang, H. Chen, Y. Li, A. Kuang, H. Yuan and G. Wu, Phys. Chem. Chem. Phys., 2015, 17, 28743–28753 RSC.
- P. Liu, J. Nisar, B. Pathak and R. Ahuja, Phys. Chem. Chem. Phys., 2013, 15, 17150–17157 RSC.
- P. Liu, J. Nisar, R. Ahuja and B. Pathak, J. Phys. Chem. C, 2013, 117, 5043–5050 CAS.
- Y. Jiang, H. Yuan and H. Chen, Phys. Chem. Chem. Phys., 2015, 17, 630–637 RSC.
- W.-J. Yin, S.-H. Wei, M. M. Al-Jassim and Y. Yan, Phys. Rev. Lett., 2011, 106, 066801 CrossRef PubMed.
- N. Meir, I. Jen-La Plante, K. Flomin, E. Chockler, B. Moshofsky, M. Diab, M. Volokh and T. Mokari, J. Mater. Chem. A, 2013, 1, 1763–1769 CAS.
- J. J. Chen, W.-K. Wang, W.-W. Li, D.-N. Pei and H.-Q. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 12671–12678 CAS.
- C. L. Yu, F. F. Gao, G. Li, R. F. Wei, R. C. Jin and Q. Z. Fan, Sep.
Purif. Technol., 2013, 120, 110–122 CrossRef CAS.
- W. Yang, Y. Wen, D. Zeng, Q. Wang, R. Chen, W. Wang and B. Shan, J. Mater. Chem. A, 2014, 2, 20770–20775 CAS.
- A. Torabi and V. N. Staroverov, J. Phys. Chem. Lett., 2015, 6, 2075–2080 CrossRef CAS PubMed.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- T. Leijtens, J. Lim, J. Teuscher, T. Park and H. J. Snaith, Adv. Mater., 2013, 25, 3227–3233 CrossRef CAS PubMed.
- J. Xiong, G. Cheng, F. Qin, R. Wang, H. Sun and R. Chen, Chem. Eng. J., 2013, 220, 228–236 CrossRef CAS.
- J. Xiong, G. Cheng, G. Li, F. Qin and R. Chen, RSC Adv., 2011, 1, 1542–1553 RSC.
- W. Fang, C. Yu, J. Li and W. Z. L. Zhu, J. Mater. Res., 2015, 30, 3125–3133 CrossRef CAS.
- G. Li, F. Qin, H. Yang, Z. Lu, H. Sun and R. Chen, Eur. J. Inorg. Chem., 2012, 2012, 2508–2513 CrossRef CAS.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- H. Li, J. Shi, K. Zhao and L. Zhang, Nanoscale, 2014, 6, 14168–14173 RSC.
- H. Li and L. Zhang, Nanoscale, 2014, 6, 7805–7810 RSC.
- F. Tian, Y. Zhang, G. Li, Y. Liu and R. Chen, New J. Chem., 2014, 39, 1274–1280 RSC.
- G. Li, Q. Fan, R. Wang, S. Xiao, H. Sun and C. Rong, J. Colloid Interface Sci., 2013, 409, 43–51 CrossRef CAS PubMed.
- A. Dash, S. Sarkar, V. N. Adusumalli and V. Mahalingam, Langmuir, 2014, 30, 1401–1409 CrossRef CAS PubMed.
- X. Lin, Z. Shan, K. Li, W. Wang, J. Yang and F. Huang, Solid State Sci., 2007, 9, 944–949 CrossRef CAS.
- K.-L. Zhang, C.-M. Liu, F.-Q. Huang, C. Zheng and W.-D. Wang, Appl. Catal., B, 2006, 68, 125–129 CrossRef CAS.
- W. Wang, F. Huang and X. Lin, Scr. Mater., 2007, 56, 669–672 CrossRef CAS.
- W. Wang, F. Huang, X. Lin and J. Yang, Catal. Commun., 2008, 9, 8–12 CrossRef CAS.
- L. Zhao, X. Zhang, C. Fan, Z. Liang and P. Han, Physica B: Condensed Matter, 2012, 407, 3364–3370 CrossRef CAS.
- Z.-Y. Zhao and W.-W. Dai, Inorg. Chem., 2014, 53, 13001–13011 CrossRef CAS PubMed.
- W. L. Huang and Q. Zhu, Comput. Mater. Sci., 2009, 46, 1076–1084 CrossRef CAS.
- W. L. Huang and Q. Zhu, Comput. Mater. Sci., 2008, 43, 1101–1108 CrossRef CAS.
- J. Henle, P. Simon, A. Frenzel, S. Scholz and S. Kaskel, Chem. Mater., 2007, 19, 366–373 CrossRef CAS.
- M. Sun, Q. Zhao, C. Du and Z. Liu, RSC Adv., 2015, 5, 22740–22752 RSC.
- H. Cheng, B. Huang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 97–99 RSC.
- X. Wu, H. Zhou, S. Gu, F. Wang, J. Liu and W. Li, RSC Adv., 2015, 5, 92769–92777 RSC.
- L. Kong, Z. Jiang, T. Xiao, L. Lu, M. O. Jones and P. P. Edwards, Chem. Commun., 2011, 47, 5512–5514 RSC.
- L. Hu, S. Dong, Q. Li, J. Feng, Y. Pi, M. Liu, J. Sun and J. Sun, J. Alloys Compd., 2015, 633, 256–264 CrossRef CAS.
- Y. I. Choi, Y.-I. Kim, D. W. Cho, J.-S. Kang, K. Leung and Y. Sohn, RSC Adv., 2015, 5, 79624–79634 RSC.
- F. Shen, L. Zhou, J. Shi, M. Xing and J. Zhang, RSC Adv., 2015, 5, 4918–4925 RSC.
- C. Yu, J. Chen, W. Zhou, L. Wei and Q. Fan, Mater. Res. Innovations, 2015, 19, 54–59 CrossRef CAS.
- S. Kang, R. C. Pawar, Y. Pyo, V. Khare and C. S. Lee, J. Exp. Nanosci., 2016, 11, 259–275 CrossRef CAS.
- X.-j. Wang, W.-y. Yang, F.-t. Li, J. Zhao, R.-h. Liu, S.-j. Liu and B. Li, J. Hazard. Mater., 2015, 292, 126–136 CrossRef CAS PubMed.
- M. Zhang, Y. Liu, L. Li, H. Gao and X. Zhang, Catal. Commun., 2015, 58, 122–126 CrossRef CAS.
- J. Xia, Y. Ge, D. Zhao, J. Di, M. Ji, S. Yin, H. Li and R. Chen, CrystEngComm, 2015, 17, 3645–3651 RSC.
- S. Yi, F. Zhao, X. Yue, D. Wang and Y. Lin, New J. Chem., 2015, 39, 6659–6666 RSC.
- S. Zhang and J. Yang, Ind. Eng. Chem. Res., 2015, 54, 9913–9919 CrossRef CAS.
- L. Sun, L. Xiang, X. Zhao, C. J. Jia, J. Yang, Z. Jin, X. Cheng and W. Fan, ACS Catal., 2015, 5, 3540–3551 CrossRef CAS.
- Z. Liu, H. Ran, B. Wu, P. Feng and Y. Zhu, Colloids Surf., A, 2014, 452, 109–114 CrossRef CAS.
- X. Chang, M. Gondal, A. Al-Saadi, M. Ali, H. Shen, Q. Zhou, J. Zhang, M. Du, Y. Liu and G. Ji, J. Colloid Interface Sci., 2012, 377, 291–298 CrossRef CAS PubMed.
- J. Zhang, J. Xia, S. Yin, H. Li, H. Xu, M. He, L. Huang and Q. Zhang, Colloids Surf., A, 2013, 420, 89–95 CrossRef CAS.
- T. B. Li, G. Chen, C. Zhou, Z. Y. Shen, R. C. Jin and J. X. Sun, Dalton Trans., 2011, 40, 6751–6758 RSC.
- F. Dong, Y. Sun, M. Fu, Z. Wu and S. Lee, J. Hazard. Mater., 2012, 219, 26–34 CrossRef PubMed.
- J. Cao, B. Xu, B. Luo, H. Lin and S. Chen, Catal. Commun., 2011, 13, 63–68 CrossRef CAS.
- L. Lin, Y. Wang, M. Huang and D. Chen, Aust. J. Chem., 2016, 69, 119–125 CAS.
- K. Lv, B. Cheng, J. Yu and G. Liu, Phys. Chem. Chem. Phys., 2012, 14, 5349–5362 RSC.
- N. Kijima, K. Matano, M. Saito, T. Oikawa, T. Konishi, H. Yasuda, T. Sato and Y. Yoshimura, Appl. Catal., A, 2001, 206, 237–244 CrossRef CAS.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 169 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. Liu, G. Chen, Z. Li and Z. Zhang, J. Solid State Chem., 2006, 179, 3704–3708 CrossRef CAS.
- M. Butler and D. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS.
- D. Ginley and M. Butler, J. Electrochem. Soc., 1978, 125, 1968–1974 CrossRef CAS.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- E. Keller and V. Krämer, Z. Naturforsch., B: J. Chem. Sci., 2005, 60, 1255–1263 CAS.
- J. Guo, S. Ouyang, P. Li, Y. Zhang, T. Kako and J. Ye, Appl. Catal., B, 2013, 134, 286–292 CrossRef.
- A. Serra, X. Domenech, C. Arias, E. Brillas and J. Peral, Appl. Catal., B, 2009, 89, 12–21 CrossRef CAS.
- J. Sheng, X. Li and Y. Xu, ACS Catal., 2014, 4, 732–737 CrossRef CAS.
- G. Liu, T. Wang, S. Ouyang, L. Liu, H. Jiang, Q. Yu, T. Kako and J. Ye, J. Mater. Chem. A, 2015, 3, 8123–8132 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.