
A T–Hg2+–T metallo-base pair-mediated dual amplification fluorescent strategy for the selective and sensitive detection of Hg2+†
Abstract
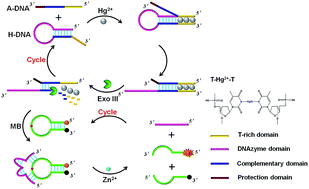
The mercuric ion is a highly toxic contaminant and causes severe harm to the environment and human health. Herein, a T–Hg2+–T metallo-base pair-mediated dual amplification fluorescent strategy was proposed for the selective and sensitive detection of Hg2+ based on a target cycle and DNAzyme cycle. First, Hg2+ selectively bound with T–T mismatches in H-DNA and A-DNA to form stable T–Hg2+–T metallo-base pairs. This initiated the strand displacement between H-DNA and A-DNA to obtain the Hg2+-mediated partial double-stranded structure, with a blunt 3′-terminus of the opened H-DNA (donated as the Hg-complex). Next, under the action of Exo III, the Hg-complex was digested to release DNAzyme, A-DNA and Hg2+. The released Hg2+ could bind with another A-DNA and H-DNA, and the target cycle started anew, eventually generating numerous DNAzymes. DNAzymes then catalyzed the cleavage of a molecular beacon (MB) to generate free fluorophores. Upon cleavage, DNAzymes were released and continuously hybridized with another MB to trigger a second DNAzyme cycle. Finally, numerous fluorophores were liberated, resulting in a significantly amplified signal. The strategy showed a good linear relationship in the range from 2.0 × 10−10 mol L−1 to 1.0 × 10−8 mol L−1, with a detection limit of 7.2 × 10−11 mol L−1. The proposed strategy exhibited remarkable selectivity towards Hg2+ against other metal ions. Furthermore, this strategy was successfully applied to detect Hg2+ in real water samples. The proposed strategy provided a reliably quantitative candidate for potential application in environmental monitoring and biotoxicity analysis.
 Please wait while we load your content...
Please wait while we load your content...

