
Direct access to aggregation-free and small intermetallic nanoparticles in ordered, large-pore mesoporous carbon for an electrocatalyst†
Yeongdong Mun‡
a,
Jongmin Shim‡a,
Kyeounghak Kimb,
Jeong Woo Hanb,
Soo-Kil Kimc,
Youngjin Yea,
Jongkook Hwanga,
Seonggyu Leea,
JongHyun Jangd,
Yong-Tae Kime and
Jinwoo Lee*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang 790-784, Republic of Korea. E-mail: jinwoo03@postech.ac.kr
bDepartment of Chemical Engineering, University of Seoul, 163 Siripdae-ro, Dongdaemun-gu, Seoul 130-742, Republic of Korea
cSchool of Integrative-Engineering, Chung-Ang University, 84 Heukseokno, Dongjak-gu, Seoul, 156-756, Republic of Korea
dFuel Cell Research Center, Korea Institute of Science and Technology (KIST), 136-791, Republic of Korea
eSchool of Mechanical Engineering, Pusan National University, Busan 609-735, Republic of Korea
First published on 30th August 2016
Abstract
An intermetallic catalyst with ordered atomic arrays has a higher electrocatalytic activity than alloy, but the high temperature required for the formation makes the particles large, resulting in low mass activity. We report the simple synthesis of small Pt-based intermetallic nanoparticles on a carbon-based ordered mesoporous support by combining block copolymer-assisted evaporation-induced self-assembly and strong metal-support interaction (SMSI). Aluminosilicate in the mesostructured wall is an SMSI agent and charge transfer from Pt to the aluminosilicate suppresses the sintering of intermetallic nanoparticles. Intermetallic PtPb and Pt3Co on carbon-based mesoporous supports are synthesized, and their particle sizes are below 5 nm even at high loading. The PtPb catalyst shows 15 times higher mass activity for formic acid oxidation than Pt/C, and the Pt3Co catalyst shows 3.25 times higher mass activity for oxygen reduction than Pt/C. This procedure can be extended to synthesize various heterogenous catalysts that require high temperature for synthesis or to operate.
Introduction
Developing a catalyst for a chemical reaction is a big issue for various science and engineering fields, with respect to controlling the reactions to achieve a desired outcome. Especially, interest in electrocatalysts has increased sharply during recent years due to the development of energy storage and conversion devices, which can function as new and renewable energy sources.1,2 Noble metals, such as platinum or palladium, are active catalysts for various electrochemical reactions. However, because of their high costs, alternatives are under development.3,4 A well-known strategy to modify the intrinsic properties and reduce the amount of noble metal is to make multicomponent catalysts, such as alloys or intermetallic compounds, with transition metals.5–12 Electrocatalysts that consist of ordered atomic arrays of intermetallic structures show higher catalytic activities than alloy catalysts that have disordered structures.5,13–16 Murray and co-workers reported the synthesis of intermetallic Pt3Zn nanocrystals, which show higher catalytic activity for methanol electrooxidation and a stronger CO poisoning tolerance than the alloy phase of Pt and Zn.17 A carbon-supported ordered intermetallic Pt3Co nanocatalyst for the oxygen reduction reaction (ORR), developed by Abruña and co-workers, has remarkably higher mass activity, specific activity, and stability than a disordered Pt3Co nanocatalyst.13They also reported promising electrocatalytic behaviors of a number of intermetallic phases toward small organic molecule oxidations. Intermetallic PtPb and PtBi phases are highly active electrocatalysts in the formic acid oxidation reaction (FOR).5 However, because of the high enthalpies of mixing during the formation of intermetallics, the synthesis procedures usually require heat-treatment at high temperatures (>500 °C), which causes the formation of large particles that have a small active area.
Thus, one of the sensible approaches to developing high-performance electrocatalysts is to synthesize small nanocatalysts (<5 nm) that have an intermetallic phase. Sun and co-workers coated an MgO protective layer on pre-synthesized disordered fcc-FePt nanoparticles to prevent them from aggregating during heating. Heating at high temperature, removing the MgO layer, and loading on a carbon support yielded an intermetallic fct-FePt nanocatalyst with high activity and stability for the ORR.16 Nazar and co-workers used the interaction between the metal precursor and sulfur to suppress sintering of the nanoparticles.14 They coated sulfur on the surface of the ordered mesoporous carbon (OMC) support before impregnation of the metal precursor. The metal particles were adsorbed strongly on the sulfur, so they did not aggregate with each other during high temperature treatment. These approaches proved the potential of small intermetallic nanoparticles as an electrocatalyst, but a more simplified process is required for practical use.
Using metal-support interactions is an interesting way to make the particles small without tiresome coating and removal of the protective layer.14 The size of the metal particles is affected considerably by the support material (generally, metal oxides, such as TiO2, Al2O3, SiO2, or ZrO2).18–20 However, in many cases, supports that interact strongly with metal usually have very low electrical conductivities. Thus, suppressing particle growth at high temperatures by strong metal-support interaction (SMSI), and providing high electrical conductivity to support an electrocatalyst, are difficult tasks to achieve simultaneously. Furthermore, the simplicity of the synthesis should be considered for a practical application.
Herein, we report a novel and simple process that suppresses the sintering of an intermetallic nanocatalyst on an OMC support at high temperature by combining SMSI and block copolymer self-assembly. Block copolymer-assisted evaporation-induced self-assembly (EISA) is a useful and simple method of synthesis of various hybrid mesoporous materials with highly ordered structures.21–29 Mesoporous materials with high surface area and controlled pore size have been utilized as promising electrode materials in energy conversion and storage devices.30–36 Previously, our group reported a synthesis method for an intermetallic PtPb nanocatalyst in ordered mesoporous carbon/silica, with poly(styrene)-block-poly(ethylene oxide) (PS-b-PEO), block copolymer-assisted EISA incorporating hydrophobic metal precursors, and a hydrophilic carbon/silica precursor.27 However, the sole use of block copolymer assembly for the synthesis of composite material still had two severe limitations. The particle size was still large (>10 nm) and the permitted loading amount of intermetallic catalyst was limited (<10 wt%). To remedy these limitations, we developed a new EISA method by incorporating an agent that strongly interacted with metal (EISA-SIM). The interacting agent in this work was aluminosilicate. By adding aluminosilicate sol to the assembly system of block copolymer, hydrophilic carbon precursor, and hydrophobic metal precursors, followed by high temperature treatment, small metal particles on an ordered mesoporous support could be obtained. Block copolymer-assisted self-assembly allowed the aluminosilicate to mix thoroughly with conductive carbon in the ordered mesoporous framework, so the mesoporous support could provide both an SMSI effect and a conductive pathway to the loaded metal particles. Intermetallic PtPb and Pt3Co nanoparticles on ordered mesoporous carbon/aluminosilicate were synthesized using an EISA-SIM process, and the size of the metal particles was below 5 nm. The small size was maintained when the loading was quite high (>20 wt%). Their electrocatalytic activities for the FOR and the ORR were superior to those of commercial and reported materials. The strong interaction between the loaded metal and aluminosilicate support was demonstrated using density functional theory (DFT) calculations and X-ray photoelectron spectroscopy (XPS). The EISA-SIM process suggests a promising way to develop small-sized and highly active catalysts that are synthesized or operate at high temperature.
Results and discussion
Small intermetallic nanoparticles were successfully incorporated in the large pores of the ordered mesoporous carbon/aluminosilicate composite by an EISA-SIM method, followed by thermopolymerization of resol at 100 °C and heat treatment at 700 °C in an H2/Ar atmosphere (Fig. 1). In a previous study, we found that the amphiphilic block copolymer, PS-b-PEO, with a high hydrophobicity/hydrophilicity contrast between the two blocks, induces microseparation of the hydrophobic PS block with hydrophobic metal precursors and the hydrophilic PEO block with aluminosilicate sol and carbon precursor resol.27 This phenomenon enabled successful confinement of metal particles in the mesopores after the final heat treatment.27 This perfect confinement of particles could not be achieved by using a commercial pluronic block copolymer.27 During thermal treatment, sp2-hybridized carbons in the PS block were partially converted to amorphous carbon, which prevents excessive growth of metal particles. Although the material in our previous work showed a higher activity and stability for the FOR than commercial materials, the size of intermetallic nanoparticles was still large (>10 nm) and the possible loading was low (<10 wt%). The large particles result in poor distribution of the noble metal, and the low metal loading (i.e. proportion) in the catalyst increases both the required amount of the support material and the electrode thickness in the membrane-electrode assembly (MEA), resulting in an increase in the mass transport resistance.37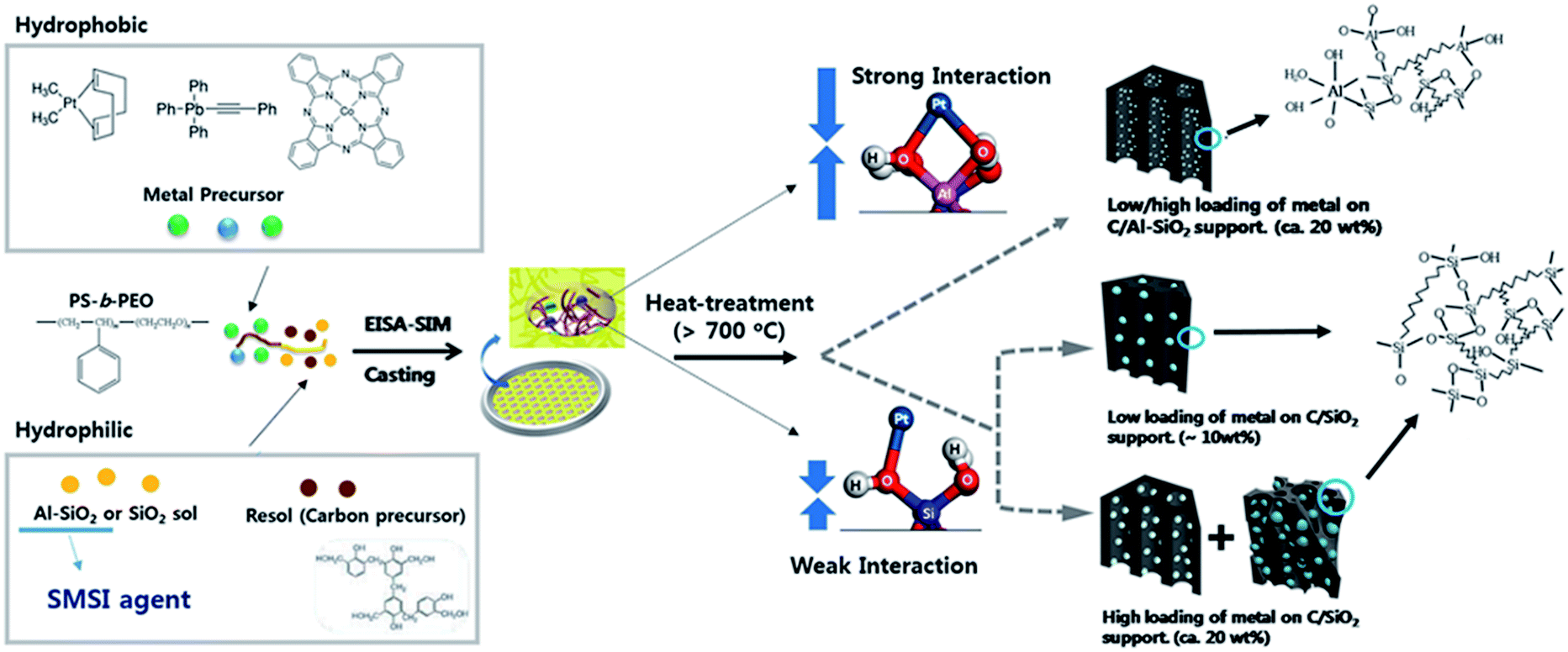 | ||
| Fig. 1 Schematic of EISA-SIM method for synthesizing metal nanoparticles within ordered mesoporous carbon/aluminosilicate (M-OMCA) and carbon/silica (M-OMCS) (M: Pt, PtPb, or Pt3Co). | ||
In this work, a decrease in the size of the nanoparticles and an increase in the metal loading were achieved simultaneously by introducing aluminosilicate into the pore wall of the mesoporous material as an agent to interact strongly with the metal. Alumina interacts strongly with Pt-group metals and induces a relatively small particle size and good dispersion of the loaded metal.38–40 Xiao and Schneider suggested a mechanism of metal-atom adsorption on the α-alumina surface, due to charge transfer and a hydrogen spillover effect, with a theoretical analysis using DFT calculations.41
Samples in this work are denoted as M-x-OMCA or M-x-OMCS; M-x-OMCA means that metal M is loaded on an ordered mesoporous carbon/aluminosilicate support with x wt%; OMCS means that the support of the sample has a carbon/silica framework, rather than carbon/aluminosilicate. The aluminosilicate prepared in this synthesis was derived from GLYMO/Al(OsBu)3 and has both tetrahedrally-coordinated and octahedrally-coordinated Al atoms.23,42 The tetrahedrally-coordinated aluminum is ascribed to Al bonds with Si via O bridges, whereas the octahedrally-coordinated Al is ascribed to aluminum oxo–hydroxo complexes, AlOx(OH)y(H2O)z.42 The composition of tetrahedral and octahedral Al sites of Pt-loaded mesoporous carbon/aluminosilicate (measured with Pt-5-OMCA) was investigated using magic-angle spinning (MAS) nuclear magnetic resonance (NMR) (Fig. 2). A high-intensity peak at 52.61 ppm is ascribed to tetrahedrally-coordinated sites, and a low-intensity peak at 1.67 ppm is ascribed to octahedrally-coordinated aluminum sites.43 Al in distorted tetrahedral sites (peak rises around 30–40 ppm) was barely observed. The ratio of the peak areas of Al at tetrahedral and octahedral sites was 93/7, and it is concluded that the majority of Al in our system is tetrahedrally-coordinated.
 | ||
| Fig. 2 27Al MAS spectrum of Pt-5-OMCA: the ratio of tetrahedral to octahedral aluminum is 93/7. | ||
To investigate the strong interaction effect between aluminosilicate and Pt-group metals in this system, mesoporous materials containing Pt nanoparticles were synthesized as a model system. To synthesize Pt-x-OMCA and Pt-x-OMCS, dimethyl(1,5-cyclooctadiene)-platinum(II) was used as a hydrophobic Pt precursor and heat treatment at 700 °C was performed under an inert atmosphere. Powder X-ray diffraction (XRD) patterns of Pt-x-OMCAs and Pt-x-OMCSs (Fig. 3a) were identified as pure Pt (JCPDS #87-0646), and transmission electron microscopy (TEM) images were captured for each compound (Fig. 3b–e). The average crystallite sizes of Pt-5-OMCA, Pt-20-OMCA, Pt-5-OMCS, and Pt-20-OMCS, calculated using the Debye–Scherrer equation44 were 3.3 nm, 3.8 nm, 14.4 nm, and 14.5 nm, respectively. The calculated Pt crystallite sizes of Pt-x-OMCA were much smaller than those of Pt-x-OMCS, regardless of the amount of Pt; this difference indicates that the interaction between Pt and the support is strongly related to the metal particle size. TEM images of Pt-x-OMCAs and Pt-x-OMCSs (Fig. 3b–e) also clearly exhibited the different particle sizes. The particle size was below 4 nm on the OMCA supports. Ordered mesoporous structures of 2-D hexagonally-arranged cylindrical channels with pore sizes from 35 to 45 nm were formed in all samples. The uniformity of the mesopores was also supported by the type-IV curves with H1-type hysteresis in N2 adsorption/desorption isotherms, and by the sharp peaks in the pore-size distributions (Fig. S1†). Surface characteristics (Table S1†) of the samples were measured using N2 physisorption. Small-angle X-ray scattering (SAXS) patterns (Fig. 4) of Pt-5-OMCA and Pt-20-OMCA showed distinct peaks at the relative scattering vector (q) positions of the first-order maxima, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31/2:41/2:71/2:121/2:131/2:211/2, which correspond to the p6mm hexagonally-arranged channel structure.44 The SAXS peaks of Pt-20-OMCA were at a lower angular position than those of Pt-5-OMCA because the former contained more hydrophobic Pt precursor, and its PS block domain swelled more than that of the latter.
:31/2:41/2:71/2:121/2:131/2:211/2, which correspond to the p6mm hexagonally-arranged channel structure.44 The SAXS peaks of Pt-20-OMCA were at a lower angular position than those of Pt-5-OMCA because the former contained more hydrophobic Pt precursor, and its PS block domain swelled more than that of the latter.
 | ||
| Fig. 3 (a) Powder XRD patterns of Pt-5-OMCA (black), Pt-20-OMCA (red), Pt-5-OMCS (blue), and Pt-20-OMCS (green). TEM images: (b) Pt-5-OMCA, (c) Pt-20-OMCA, (d) Pt-5-OMCS, and (e) Pt-20-OMCS. | ||
 | ||
| Fig. 4 SAXS patterns: (a) Pt-20-OMCA (red) and Pt-5-OMCA (black); (b) Pt-20-OMCS (red) and Pt-5-OMCS (black). The asterisks indicate the first order peaks. | ||
Pt particles on OMCS supports grew to larger than 10 nm, even when the loading was as low as 5 wt% (Fig. 3d and e), due to the insufficient strength of the Pt metal adsorption on OMCS. The TEM images and pore size distributions show that the ordered mesostructures were preserved in both Pt-20-OMCS and Pt-5-OMCS, but the structure was less ordered in Pt-20-OMCS than in Pt-5-OMCS, and in some parts of Pt-20-OMCS, the structure was destroyed because some particles grew larger than the mesopore diameter (Fig. S2a†). The peaks in the SAXS pattern were much less distinct in Pt-20-OMCS than in the other samples, except for the first-order peak (Fig. 4b); this difference indicates poor long-range order in Pt-20-OMCS due to the partial destruction of the mesostructure.
To verify the strong binding of Pt metal on aluminosilicate and its effect on the Pt particle size more clearly, theoretical modeling and experimental measurement were performed with DFT calculations and XPS measurement, respectively. DFT calculation enabled a qualitative comparison of the Pt adsorption strength on a pure SiO2 surface with that on Al-doped SiO2 (aluminosilicate). As revealed by the MAS spectrum of 27NMR of Pt-5-OMCA, because most aluminum is tetrahedrally coordinated (Fig. 2), the DFT calculation was conducted using SiO2 of β-cristobalite (001) with Al doping in tetrahedral sites as the model. It was found that a Pt atom preferentially binds to the top site of a surface oxygen atom on pure SiO2 (Fig. 5a), whereas on Al-doped SiO2, the Pt adsorbed on the bridge site between the surface O atoms (Fig. 5b). On both pure and Al-doped SiO2, the adsorption of a Pt atom was exothermic, with negative adsorption energy (Table 1). Al-doping on the silica support increased the strength of Pt adsorption significantly by 2.22 eV. To elucidate the difference in the adsorption between the pure and the Al-doped SiO2 surfaces, the electronic structural changes were analyzed. Charge density difference plots for Pt adsorbed on both surfaces (Fig. 5c and d) were constructed from the difference between the charge densities of a Pt atom and the surface fixed in its adsorbed geometry, and the charge density of Pt adsorbed on each surface. Charge depletion near the Pt atom was significant for the Al-doped SiO2, whereas an accumulation of charge was observed near Pt on the pure SiO2 (Fig. 5c and d and Table 1). To quantify the charge transfer between the adsorbed Pt and the surface, we performed Bader charge analysis. For the Al-doped SiO2, a charge of 0.34e− was shifted from the Pt to the surface. For the pure SiO2, however, a charge of 0.12e− was shifted to the adsorbed Pt from the surface. These results suggest that the doped Al atom has electron-accepting empty states to attract electron density, thereby causing electron withdrawal from the adsorbed Pt atom; this causes the adsorbed Pt to be partially cationic. Because a large charge transfer leads to strong chemisorption, the Bader charge analysis indicates that Pt is adsorbed more strongly to aluminosilicate than to pure silica.
 | ||
| Fig. 5 Side views of fully hydroxylated surface and the associated Pt atom adsorption sites: (a) pure SiO2 and (b) Al-doped SiO2 surface. Charge density difference on isosurfaces of Pt-adsorbed (c) pure SiO2 and (d) Al-doped SiO2. Blue and yellow represent charge depletion and accumulation, respectively. | ||
| Pt adsorption energy (eV) | |
|---|---|
| a Charge difference on the atom with the asterisk was calculated based on Bader analysis. | |
| Pt–SiO2 | −1.53 |
| Pt–Al_SiO2 | −3.75 |
The result from XPS (Fig. S3†) strongly supports the explanation suggested by the DFT calculation. The Pt 4f7/2 spectra were deconvoluted to peaks that correspond to Pt(0), Pt(II), and Pt(IV); the proportion of oxidized platinum (Pt(II), Pt(IV)) was much higher in Pt-5-OMCA than in Pt-5-OMCS. This observation can be attributed to the high charge depletion around Pt on the aluminosilicate, as discovered by the calculation of the charge density and reported previously.39,45
EISA-SIM was used to synthesize small and highly-dispersed Pt-based intermetallic nanoparticles on an OMCA support. The target materials to be loaded on the ordered mesoporous support were intermetallic PtPb for the FOR and Pt3Co for the ORR. The target mass ratio of the final carbon:aluminosilicate and carbon:silica was fixed as 6:4. Dimethyl-(1,5-cyclooctadiene)-platinum(II), triphenyl-(phenylethynyl)-lead, and cobalt(II) phthalocyanine were used as hydrophobic Pt, Pb, and Co precursors, respectively. The weight ratio (Table S2†) of metal nanoparticles for the final composite after heat treatment was determined by ICP spectroscopy. Intermetallic Pt3Co-containing catalysts were characterized after using mild HF etching to remove aluminosilicate or silica in the mesoporous support to enhance the electrical conductivity of the support framework. Removing the insulative aluminosilicate or silica framework could facilitate the electrocatalysis of Pt3Co nanoparticles by increasing the electrical conductivity of the support and by generating micropores that facilitate the approach of the reactant to the metal particles. After etching, the Pt/Co atomic ratio was still maintained as ∼3 (Table S2†). This may be due to the formation of a protective platinum monolayer shell, which is formed at the surface of the Pt-transition metal alloy or intermetallic at high temperatures. The catalysts obtained by removing aluminosilicate or silica from Pt3Co-x-OMCA and Pt3Co-x-OMCS were denoted as Pt3Co-x-OMC(A) and Pt3Co-x-OMC(S), respectively. However, in the case of PtPb, because the etching process diminished the electrocatalytic activity, the electrocatalytic activities of the PtPb samples were tested without removal of aluminosilicate. The vulnerability of PtPb nanoparticles under etching conditions may be attributed to the relatively large exposure of secondary metal Pb, which is much more easily oxidized than platinum.46
XRD patterns (Fig. 6a and b) and electron microscopy images (Fig. 6c–h) clearly showed that the support containing aluminosilicate can suppress the sintering of Pt-based intermetallic nanoparticles. The XRD pattern of PtPb-20-OMCA matched perfectly with the reference pattern of the PtPb intermetallic compound (JCPDS #06-0374), and the single phases of Pt or Pb were not observed. The average crystallite size was 4.4 nm in PtPb-20-OMCA and 13.1 nm in PtPb-9-OMCS (Fig. 6a). Despite high-temperature heat treatment (700 °C) and high loading, PtPb on the OMCA support was not sintered severely, whereas the PtPb-9-OMCS had a large particle size with an even lower loading amount than PtPb-20-OMCA. The diffraction patterns for Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) showed peaks at 2θ = 23°, 33°, 53°, and 58°, which are well-matched to the (100), (110), (210), and (211) planes of an ordered intermetallic Pt3Co phase (JCPDS #29-0499), even after the etching process (Fig. 6b). The average particle size from the XRD pattern of Pt3Co-33-OMC(A) was much smaller (3.5 nm) than that of Pt3Co-31-OMC(S) (9.6 nm).
 | ||
| Fig. 6 Powder XRD patterns: (a) PtPb-9-OMCS (blue) and PtPb-20-OMCA (red); (b) Pt3Co-33-OMC(A) (green) and Pt3Co-31-OMC(S) (black). Vertical lines are the reference peak positions of PtPb (JCPDS #06-0374) and Pt3Co (JCPDS #29-0499), respectively. SEM images: (c) PtPb-20-OMCA; (d) Pt3Co-33-OMC(A). TEM images: (e) PtPb-20-OMCA; (f) Pt3Co-33-OMC(A). SEM images: (g) PtPb-20-OMCS; (h) Pt3Co-31-OMC(S). | ||
Structural features were revealed by electron microscopy images (Fig. 6c–h). Small PtPb and Pt3Co intermetallic nanoparticles on OMCA supports were homogeneously distributed without aggregation in the ordered mesopores. The pore sizes of the ordered mesoporous supports were 30 to 35 nm, as also shown in the pore size distributions (Fig. S4†). The BET surface area of PtPb-20-OMCA is similar to those of Pt-x-OMCA materials. Pt3Co-33-OMC(A) has a larger surface area than the other samples because micropores are generated by etching aluminosilicate (Table S3†). Despite the high loading of nanoparticles, the SAXS patterns accorded with p6mm phases (Fig. S5†); therefore, the ordered mesostructures of the OMCA supports were well-maintained without a structural collapse after high-temperature heat treatment at 700 °C. When the same amount of metal was loaded on the OMCS support, the ordered structure was disrupted in some parts (Fig. S2b and c†), and a less ordered hexagonal structure was dominant (Fig. 6g and h); this conclusion is corroborated by a reduction in the sharpness of the SAXS peaks (Fig. S5†). This observation suggests that the strong interaction between the metals and support containing aluminosilicate has an important role in the synthesis of well-dispersed small intermetallic nanoparticles on the highly ordered mesoporous support.
The abilities of the synthesized intermetallic nanoparticles to electrocatalyze the FOR and the ORR were characterized. Rotating-disk-electrode voltammograms (Fig. 7a) were obtained for the FOR of PtPb-20-OMCA, PtPb-9-OMCS, and commercial Pt/C 10 wt% in 0.5 M formic acid and 0.1 M H2SO4 solution. The oxidation current was normalized to the weight of the metal contained in each catalyst and presented as the mass activity in A mgmetal−1. PtPb-20-OMCA, PtPb-9-OMCS, and Pt/C had onset potentials of −0.2, −0.19, and −0.03 V, respectively. The mass activity value at 0.2 V (2000 rpm) for the FOR of PtPb-20-OMCA was 2.07 A mgmetal−1, which is 15 times higher than that of Pt/C (0.14 A mgmetal−1). Although PtPb-20-OMCA had a higher loading amount of catalyst than PtPb-9-OMCS, the mass activity of PtPb-20-OMCA at 0.2 V was much higher than that of PtPb-9-OMCS (1.34 A mgmetal−1). Compared to previously-reported PtPb on carbon testing under similar conditions at 0.2 V (vs. Ag/AgCl), our PtPb-20-OMCA catalyst exhibited one of the highest electrocatalytic activities for the FOR (Table S4†). The superior catalytic activity of PtPb-20-OMCA for FA oxidation should be attributed to the large surface area of the small and well-dispersed particles. Anodic FA oxidation profiles were obtained at steady-states (Fig. 7b). For Pt/C, the maximum current of the second oxidative peak at 0.73 V, which corresponds to oxidation of adsorbed CO, was much higher than the first maximum current at 0.38 V; this difference means that oxidation of FA on Pt followed the CO-mediated dehydration path.47,48 For PtPb-20-OMCA and PtPb-9-OMCS, the single anodic peaks were at lower potentials; this observation means that the desirable dehydrogenation path was more favorable than the dehydration pathway on PtPb.47,48 A CO stripping test was conducted to investigate the catalysts' tolerance for CO, which could be generated with small amounts during the reaction (Fig. 7c and d). A CO oxidation peak was shown clearly around 0.56 V in the CV curve of Pt/C, obtained after the CO adsorption process. In the curve for PtPb-20-OMCA, a totally different adsorption property was observed. There was no distinct oxidation peak of CO, and only capacitive behaviour, which was derived from the electric double-layer reaction on the mesoporous carbon surface, and oxidation/reduction of small amounts of oxygen functional groups of mesoporous carbon, were observed as described in a previous report about intermetallic PtPb.49 This result indicates the low affinity of intermetallic PtPb for CO, and this low affinity is appropriate to catalyze the FOR through the dehydrogenation path, and to prevent CO poisoning during the reaction.
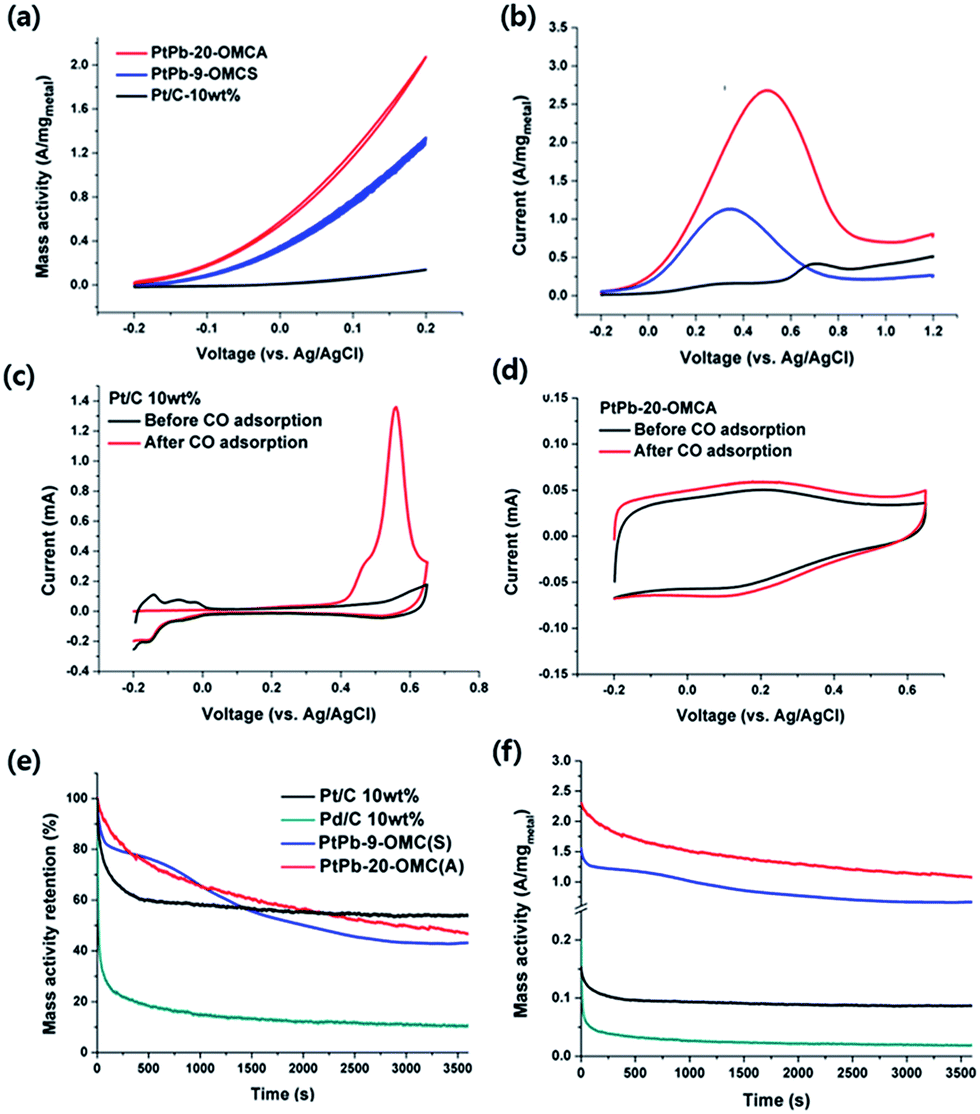 | ||
| Fig. 7 Electrochemical characterization of FOR: (a) rotating-disk electrode (10 mV s−1 and 2000 rpm); (b) steady-state voltammograms (anodic scan, 10 mV s−1). CO stripping voltammograms: (c) Pt/C 10 wt%; (d) PtPb-20-OMCA. Chronoamperometry for FOR at 0.3 V (vs. Ag/AgCl): (e) current drops; (f) current retentions; (see also Fig. S6†). All experiments were performed in 0.5 M formic acid and 0.1 M H2SO4. | ||
In addition to the high electrocatalytic activity, PtPb-20-OMCA showed a high durability for the FOR in chronoamperometry (Fig. 7e and f). The high mass activity of PtPb-20-OMCA was maintained as stably as that of Pt/C and state-of-the-art catalysts, which were reported recently (Fig. 7e and f, and Table S5†).
After performing chronoamperometry at 0.3 V for 20000 seconds, 20.5% of the initial mass activity was retained; this percentage is similar to the durability of large PtPb particles on OMCS (Fig. S6†).27 Decreasing the particle size of PtPb intermetallic particles both increased their catalytic activity significantly, and granted comparable durability to that of large PtPb particles.
The intermetallic Pt3Co nanocatalyst in an OMC support (i.e., Pt3Co-33-OMC(A)) was investigated as an electrocatalyst for the ORR. CV curves (Fig. 8a) and ORR polarization curves (Fig. 8b) were obtained with Pt3Co-33-OMC(A) and the reference group of Pt3Co-31-OMC(S) and commercial Pt/C (40 wt%, Johnson-Matthey Co.). The properties of the catalysts under the conditions of the ORR activity test, including electrochemical surface area (ECSA), mass activity, and specific activity, were calculated based on eqn (S1)–(S4),† and are summarized in Table 2.
 | ||
| Fig. 8 Electrochemical characterization for ORR: (a) cyclic voltammograms; (b) ORR polarization curves in 0.1 M HClO4 solution. (c) The size distribution nanoparticles in Pt3Co-33-OMC(A) (obtained from the selected area of the dark-field scanning transmission electron microscopy (DF-STEM) image). | ||
| Samples | ECSA (Hupd) (m2 gcatal−1) | Mass activity (mA mgcatal−1) | Specific activity (μA cmcatal−2) |
|---|---|---|---|
| Pt/C (40 wt%) | 66 | 105 | 159 |
| Pt3Co-31-OMC(S) | 32 | 215 | 671 |
| Pt3Co-33-OMC(A) | 83 | 342 | 412 |
The hydrogen underpotential-deposition (Hupd) peak in the CV curve of Pt3Co-33-OMC(A) indicates that the extremely small particles gave a larger electrochemical surface area (ECSA) than commercial Pt/C, which has a catalyst particle size of 2–3 nm (Fig. 8a and Table 2). On the other hand, Pt3Co-31-OMC(S) had a much smaller ECSA than Pt3Co-33-OMC(A) (Fig. 8a).
A surface atom of the Pt3Co nanocatalyst with the L12 intermetallic phase has a lower d-band centre than a pure Pt surface; this weakens the binding of the oxygen-containing intermediate of the ORR and speeds the reaction kinetics;13,50,51 therefore, the peaks of oxide reduction in the CV curves for Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) were slightly shifted to the positive direction as compared to Pt/C; this shift indicates that the surfaces of Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) are less oxophilic than that of Pt/C. In CV curves obtained in the O2-purged electrolyte, the positive shift of the Pt-oxide reduction peak position was observed more clearly in curves of Pt3Co-33-OMC(A) (0.860 mV vs. RHE) and Pt3Co-31-OMC(S) (0.874 mV) as compared to the peak in the CV curve for Pt/C (0.826 mV) (Fig. S7†). The polarization curves clarify the ORR kinetics on the catalysts (Fig. 8b). Half-wave potentials (E1/2) of Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) were positively shifted as compared to Pt/C. Pt3Co-33-OMC(A) had 3.25 times higher mass activity and 2.59 times higher specific activity than did Pt/C (Table 2); these increases can be ascribed to the downshifted d-band centre of the intermetallic phase of Pt3Co and the large catalytic surface area. But, the specific activity of Pt3Co-33-OMC(A) is lower than that of Pt3Co-31-OMC(S). We attributed this to the particle size of Pt3Co-33-OMC(A) being too small.52,53 A decrease in the size of nanoparticles results in an increase in the exposure of low-coordinated atoms that bind strongly to oxygen-containing species, thus making the ORR kinetics slower.52,53 The particle size distribution was obtained by measuring the diameters of 100 particles in the white inset box of the DF-STEM image in Fig. 8c, which was obtained to differentiate the crystalline metal particles from the amorphous support. A large proportion of metal particles of Pt3Co-33-OMC(A) had a very small size (<2 nm) (Fig. 8c). However, Pt3Co-31-OMC(S) had a higher specific activity than Pt3Co-33-OMC(A). Considering that the dominant particle shapes of Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) are nearly spherical, this high specific activity can be attributed to the d-band centre downshift and the relatively large size of Pt3Co intermetallic particles, with fewer low-coordinated atoms on the surface.52,53 Due to the high specific activity of Pt3Co-31-OMC(S), which is 4.22 times higher than that of Pt/C, the current density measured in the kinetic region (normally, at 0.9 V) of Pt3Co-31-OMC(S) was higher than that of Pt/C in spite of the low ECSA.
It was also revealed that the durability of the catalyst was affected strongly by the particle size, as revealed in previous studies.54 We evaluated the durability of three catalysts by comparing the mass activities at 0.9 V, before and after 3000 and 10000 potential cycles (0.6–1.1 V in Ar-purged 0.1 M HClO4, Fig. S8 and Table S6†). Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) showed much higher durability than Pt/C, due to the ordered atomic array of the intermetallic phase and the confinement effect of the mesopores. The mass activity retention of Pt3Co-31-OMC(S) was especially high during the potential cycling (84% after 3000 cycles, and 65% after 10000 cycles) due to the stable large particles. The mass activity retention of Pt3Co-33-OMC(A) (72% after 3000 cycles, and 59% after 10000 cycles) was not as high as that of Pt3Co-31-OMC(S) due to the larger fraction of low-coordinated atoms on the surface. Because of the high oxophilicity of the low-coordinated atoms on the particle surface, they are easily oxidized and dissolved from the particle, resulting in a decrease in durability. Nevertheless, the mass activity of Pt3Co-33-OMC(A) after 10000 potential cycles (202 mA mgcatal−1 at 0.9 V) was still similar to the original mass activity of Pt3Co-31-OMC(S) (215 mA mgcatal−1), and was 1.92 times higher than the original mass activity of Pt/C, due to the well-dispersed and small intermetallic particles of the original catalyst. The durability of the two intermetallic catalysts is also revealed by TEM images of catalysts obtained after durability testing (Fig. S9†). After a 10000 cycle durability test, many deformed particles and aggregates are found in Pt/C. On the other hand, only a slight increase in the metal catalyst particle size was observed in Pt3Co-33-OMC(A), and the shape and size of metal particles in Pt3Co-31-OMC(S) was almost unchanged. We attribute this high durability of Pt3Co-33-OMC(A) and Pt3Co-31-OMC(S) to the ordered atomic array of intermetallic particles and the confinement effect on particles in mesopores.
The effect of the particle size of intermetallic Pt3Co was also observed, as one of the adsorption properties of the catalyst surface, and was found by the comparison of ECSA from CO stripping (ECSACO) and the Hupd (ECSAHupd) (Fig. S10 and Table S7†). A smooth Pt-skin layer, which is formed on the surface of Pt-alloy or intermetallic at high temperature, and consists of densely-packed high-coordinated atoms, suppresses H adsorption on the catalyst surface, so ECSACO becomes 1.5 times larger than ECSAHupd.10,55 The ratio of ECSACO/ECSAHupd was 1.50 for Pt3Co-31-OMC(S), which indicates Pt-skin layer formation on Pt3Co particles during the heat treatment (Fig. S10b†). But, the ratio for Pt3Co-33-OMC(A) was 1.12, which was similar to that for Pt/C (Fig. S10a and c†). This is because the suppression of H adsorption on Pt3Co particles was not significant, due to the dominant exposure of the low-coordinated atoms, which adsorb small adsorbates strongly. In future work, we aim to tune the particle size and optimize both the ECSA and the specific activity.
Conclusions
Small intermetallic nanoparticles on OMC supports were successfully synthesized using the newly-developed EISA-SIM method assisted by a block copolymer and an aluminosilicate interacting agent. This is a simple ‘one-pot’ process. A strong SMSI effect, derived from charge transfer between the Pt metal and aluminosilicate, was shown clearly by DFT calculations. The desired morphology of the small intermetallic nanoparticles in the large and well-ordered mesopores was well maintained, even with a high loading of metal. As expected, the small PtPb and Pt3Co intermetallic nanocatalysts showed high mass activities for the FOR and the ORR, respectively, showing the potentials of this process for practical utilization. We expect that the EISA-SIM method can be extended to the synthesis of various noble-metal-based heterogeneous catalysts that require high temperatures to operate or to be synthesized, in addition to intermetallic compounds.Author contribution
Y. Mun and J. Shim synthesized materials and carried out material characterization. Y. Mun carried out the electrochemical characterization. K. Kim and J. Han did DFT calculations and helped to write this manuscript. S. Kim, J. Jang, and Y. Kim assisted with the electrochemical characterization. Y. Ye and S. Lee assisted with the material analysis. J. Hwang synthesized the block copolymer used in experiments. Y. Mun, J. Shim, and J. Lee wrote the manuscript with the help and advice of all the authors. J. Lee designed the system and supervised the research.Acknowledgements
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean Government (MEST) (NRF-2015M1A2A2056557), and the Basic Research Laboratory program (NRF-2016R1A4A1010735). This research was also supported by a grant from the Korea CCS R&D Center (KCRC) funded by the Korea government (NRF-2014M1A8A1049349). This work was supported by the New & Renewable Energy Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resources from the Ministry of Trade, Industry & Energy, Republic of Korea. (No. 20153010041750).Notes and references
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef CAS.
- P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vázquez-Alvarez, A. C. D. Angelo, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2004, 126, 4043–4049 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC.
- R. E. Cable and R. E. Schaak, J. Am. Chem. Soc., 2006, 128, 9588–9589 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- S.-I. Choi, S. Xie, M. Shao, J. H. Odell, N. Lu, H.-C. Peng, L. Protsailo, S. Guerrero, J. Park, X. Xia, J. Wang, M. J. Kim and Y. Xia, Nano Lett., 2013, 13, 3420–3425 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed.
- J. C. Bauer, X. Chen, Q. Liu, T.-H. Phan and R. E. Schaak, J. Mater. Chem., 2008, 18, 275–282 RSC.
- J. Kim, Y. Lee and S. Sun, J. Am. Chem. Soc., 2010, 132, 4996–4997 CrossRef CAS PubMed.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 CrossRef CAS PubMed.
- V. N. Kalevaru, A. Benhmid, J. Radnik, M. M. Pohl, U. Bentrup and A. Martin, J. Catal., 2007, 246, 399–412 CrossRef CAS.
- J.-H. Oh, J. Bae, S.-J. Park, P. K. Khanna and K.-W. Jun, Catal. Lett., 2009, 130, 403–409 CrossRef CAS.
- E. N. Ntainjua, A. F. Carley and S. H. Taylor, Catal. Today, 2008, 137, 362–366 CrossRef.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066–16072 CrossRef CAS PubMed.
- J. Lee, M. M. C. Orilall, S. C. Warren, M. Kamperman, F. J. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS PubMed.
- P. F. W. Simon, R. Ulrich, H. W. Spiess and U. Wiesner, Chem. Mater., 2001, 13, 3464–3486 CrossRef CAS.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu, X. Wu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS PubMed.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S.-K. Kim, T.-H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870–6881 CrossRef CAS PubMed.
- E. Kang, H. Jung, J.-G. Park, S. Kwon, J. Shim, H. Sai, U. Wiesner, J. K. Kim and J. Lee, ACS Nano, 2011, 5, 1018–1025 CrossRef CAS PubMed.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- J. Hwang, S. H. Woo, J. Shim, C. Jo, K. T. Lee and J. Lee, ACS Nano, 2013, 7, 1036–1044 CrossRef CAS PubMed.
- C. Jo, J. Hwang, H. Song, A. H. Dao, Y.-T. Kim, S. H. Lee, S. W. Hong, S. Yoon and J. Lee, Adv. Funct. Mater., 2013, 23, 3747–3754 CrossRef CAS.
- M. C. Orilall, F. Matsumoto, Q. Zhou, H. Sai, H. D. Abruña, F. J. DiSalvo and U. Wiesner, J. Am. Chem. Soc., 2009, 131, 9389–9395 CrossRef CAS PubMed.
- Y. Mun, C. Jo, T. Hyeon, J. Lee, K.-S. Ha, K.-W. Jun, S.-H. Lee, S.-W. Hong, H. I. Lee, S. Yoon and J. Lee, Carbon, 2013, 64, 391–402 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177–2178, 10.1039/A906872D.
- J. Park, Y.-S. Jun, W. Lee, J. A. Gerbec, K. A. See and G. D. Stucky, Chem. Mater., 2013, 25, 3779–3781 CrossRef CAS.
- M. Lee, M. Uchida, D. A. Tryk, H. Uchida and M. Watanabe, Electrochim. Acta, 2011, 56, 4783–4790 CrossRef CAS.
- E. Xue, K. Seshan and J. R. H. Ross, Appl. Catal., B, 1996, 11, 65–79 CrossRef CAS.
- R. Burch and A. Ramli, Appl. Catal., B, 1998, 15, 63–73 CrossRef CAS.
- M.-Y. Kim, J.-H. Park, C.-H. Shin, S.-W. Han and G. Seo, Catal. Lett., 2009, 133, 288–297 CrossRef CAS.
- L. Xiao and W. F. Schneider, Surf. Sci., 2008, 602, 3445–3453 CrossRef CAS.
- M. Templin, A. Franck, A. Du Chesne, H. Leist, Y. Zhang, R. Ulrich, V. Schädler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS PubMed.
- M. Templin, U. Wiesner and H. W. Spiesss, Adv. Mater., 1997, 9, 814–817 CrossRef CAS.
- A. Krawitz, Introduction to diffraction in materials science and engineering, Wiley, New York, 2001 Search PubMed.
- S. S. Soni, G. Brotons, M. Bellour, T. Narayanan and A. Gibaud, J. Phys. Chem. B, 2006, 110, 15157–15165 CrossRef CAS PubMed.
- S. D. Jackson, J. Willis, G. D. McLellan, G. Webb, M. B. T. Keegan, R. B. Moyes, S. Simpson, P. B. Wells and R. Whyman, J. Catal., 1993, 139, 191–206 CrossRef CAS.
- D. R. Blasini, D. Rochefort, E. Fachini, L. R. Alden, F. J. DiSalvo, C. R. Cabrera and H. D. Abruña, Surf. Sci., 2006, 600, 2670–2680 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 CrossRef CAS PubMed.
- F. Matsumoto, C. Roychowdhury, F. J. DiSalvo and H. D. Abruña, J. Electrochem. Soc., 2008, 155, B148–B154 CrossRef CAS.
- C. Wang, M. Chi, D. Li, D. Strmcnik, D. F. van der Vliet, G. Wang, V. Komanicky, K.-C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, B. B. Blizanac, M. Arenz, V. R. Stamenkovic, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2005, 109, 14433–14440 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- S. G. Rinaldo, J. Stumper and M. Eikerling, J. Phys. Chem. C, 2010, 114, 5773–5785 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods N2 physisorption results, elemental compositions, electrochemical surface area, XPS data, SAXS data of samples. See DOI: 10.1039/c6ra14861a |
| ‡ Y. Mun and J. Shim contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |