
Controlled electrochemical and electroless deposition of noble metal nanoparticles on graphene†
Abstract
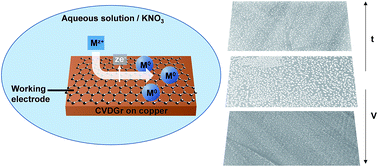
Electrodeposition of nanoparticles on graphene is described, providing a convenient approach for making graphene–nanoparticle composites as well as insight into the electrochemical activity of graphene. To avoid complications due to surface contamination, chemical vapour deposition grown graphene was used directly on its copper growth substrate. We identify and isolate two nanoparticle growth processes for both silver and palladium deposition: electroless deposition that appears to occur preferentially at defects and edges next to the underlying copper, and conventional electrodeposition that occurs uniformly across the graphene surface. We show that control over electrodeposited nanoparticle size and number density can be achieved through varying electrodeposition conditions. The resultant nanoparticles are homogeneously dispersed across the graphene surface, suggesting that here both edge-plane and basal-plane graphene sites are electroactive. These results demonstrate that, as with other carbon nanostructures, electrodeposition is a powerful and flexible tool for forming functional composites with graphene.
 Please wait while we load your content...
Please wait while we load your content...

