
Atmospheric oxidation mechanism of OH-initiated reactions of diethyl ether – the fate of the 1-ethoxy ethoxy radical†
Abstract
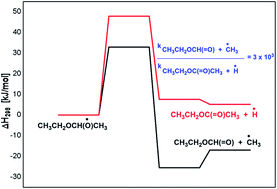
The oxidation of diethyl ether (DEE) by hydroxyl radical is studied by means of density functional theory and coupled cluster methods. The OH-initiated reactions were found to proceed by H-atom abstraction from –CH3 or –CH2 groups of DEE, in which the latter reaction is found to be more favourable than the former. The secondary reactions associated with the peroxy radical and the following alkoxy radical chemistry of DEE is explored in detail. The 1-ethoxy ethoxy radical resulting from the peroxy radical chemistry of DEE undergoes –CH3 as well as H-atom elimination reactions leading to the formation of ethyl formate and ethyl acetate in the respective reactions, where both reactions are kinetically competitive. The next significant reaction in the 1-ethoxy ethoxy radical decomposition is its reaction with O2, where ethyl acetate and HO2 are formed. The decomposition of the 1-ethoxy ethoxy radical via C–O bond cleavage is less feasible when compared to the above reactions. The reactions of 1-ethoxy ethoxy radicals with nitrates are largely thermodynamic driven, but poorly kinetic driven. The calculated results show that the major product from the oxidation chemistry of DEE is ethyl formate, followed by the formation of ethyl acetate in minor quantities. The results obtained from the current theoretical study are in excellent agreement with the available literature.
 Please wait while we load your content...
Please wait while we load your content...

