
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Heterotriangulene chromophores with 4-pyridyl anchors for dye-sensitized solar cells†
Ute
Meinhardt
a,
Fabian
Lodermeyer
b,
Tobias A.
Schaub
a,
Andreas
Kunzmann
b,
Pavlo O.
Dral
c,
Anna Chiara
Sale
a,
Frank
Hampel
a,
Dirk M.
Guldi
*b,
Ruben D.
Costa
*b and
Milan
Kivala
*a
aDepartment of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen-Nürnberg, Henkestraße 42, 91054 Erlangen, Germany. E-mail: milan.kivala@fau.de
bDepartment of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de; ruben.costa@fau.de
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
First published on 20th July 2016
Abstract
A series of dimethylmethylene-bridged N-heterotriangulenes decorated with one, two, and three electron-withdrawing 4-pyridyls were synthesized. Their photophysical and electrochemical characteristics were examined and their successful application in n-type TiO2- and ZnO-based dye-sensitized solar cells demonstrated the ability of the 4-pyridyl moiety to act as an anchor.
Starting with the pioneering work by Grätzel and co-workers, dye-sensitized solar cells (DSSCs),1 turned into frontrunners towards the development of environmentally benign energy sources.2 One of the biggest challenges in the field, that is, academic and industrial research, constitutes the development of metal-free organic sensitizers as an alternative to ruthenium-based dyes. Here, their low-cost syntheses, their high molar extinction coefficients, and the possibility to efficiently control their electronic and molecular structures through molecular design are important assets, which have fueled the research in this field during recent years.3 Most of the organic dyes with an electron donor–π-bridge–acceptor (D–π–A) architecture feature broad and intense absorptions in the visible region of the solar spectrum as a consequence of an intramolecular charge-transfer (ICT).4 Numerous types based on organic electron donors, such as indoline,5 thiophene,6 dithiafulvene,7 4H-pyran-4-ylidene,8 in general, and, triphenylamine,9,10 in particular, have been developed and applied in DSSCs. Notably, dimethylmethylene-bridged triphenylamine (DTPA), so-called N-heterotriangulene,11 has only recently been employed as an electron donor part in dyes with enhanced photovoltaic performances.12 Vital for the performance is the planarization of the originally propeller-shaped triphenylamine.
The acceptor part most often consists of a carboxylic or cyanoacrylic acid as anchor to ensure efficient electronic communication through the formation of a strong bidentate linkage with Brønsted acid sites at the TiO2 surface.3,4,13 Recently, an electron-withdrawing 4-pyridyl has been reported by Ooyama and co-workers as an alternative anchor.14 It enables efficient electron injection upon forming strong coordinative bonds with the Lewis acidic sites at the TiO2 surface.15 Since then, pyridyl linkers have been combined with a variety of electron donors such as carbazole,16 porphyrins,17 boron-dipyrromethene,18 tetrahydroquinoline,19 and triphenylamine.20
To our surprise, organic sensitizers combining N-heterotriangulene donors with pyridyl anchoring moieties have not been reported to date. As part of our ongoing research on functional molecules and materials based on N-heterotriangulenes,21 we have designed and synthesized a series of dimethylmethylene-bridged N-heterotriangulenes decorated with one, two, and three electron-withdrawing 4-pyridyl anchoring groups. The resulting electron donor–acceptor dyes were studied regarding their photophysical and electrochemical properties, as well as their integration into DSSCs featuring TiO2 and ZnO electrodes.
For the synthesis of mono-1, bis-2, and tris-pyridyl N-heterotriangulene 3, the corresponding brominated precursors, prepared by treatment of the parent DTPA with an appropriate amount of N-bromosuccinimide,22,23 were subjected to Pd-catalyzed Suzuki–Miyaura cross-coupling reaction with 4-pyridylboronic acid under microwave irradiation (Fig. 1; for experimental details, see ESI†). The target compounds 1–3 were isolated in good yields between 55% (1), and 86% (3) as orange solids. They are stable under ambient conditions and soluble in common organic solvents. The chemical identity of 1–3 was confirmed by 1H and 13C NMR spectroscopy, high resolution mass spectrometry, and elemental analysis (see ESI†).
 | ||
| Fig. 1 Chemical structures of N-heterotriangulene (DTPA) and mono-1, bis-2, and trispyridyl substituted dyes 3; in brackets, the yields of the Suzuki–Miyaura cross-coupling reaction between 4-pyridylboronic acid and the corresponding brominated DTPA are shown. | ||
Single crystals of 2 suitable for X-ray crystallographic analysis were grown by slow evaporation of a solution of 2 in CH2Cl2/MeOH at room temperature. While the central ‘C3N’ amine motif appears to be virtually planar with the sum of C–N–C angles of 359.5°, the flexibility of the C(sp3)-bridges enables slightly bent molecular geometry in the solid state in analogy to the parent DTPA (Fig. 2a).22 Taking the moderate torsion angles of 29.1° and 43.8° between the two pyridine rings of 2 and the N-heterotriangulene framework (defined by the plane passing through C1/C13/C20/N1) into account, considerable electronic communication between the electron-donating N-heterotriangulene and the electron-withdrawing 4-pyridyls can be anticipated. In the crystal packing, efficient π–π stacking is prevented by the tetrahedral dimethylmethylene bridges. Such a steric hindrance leads to a herringbone arrangement with alternating distances of the individual layers of 3.79 and 4.83 Å and a tilt angle of 79.1° between individual molecules in adjacent stacks (Fig. 2b). The observed packing motif is predominantly governed by multiple C(sp3)–H/π interactions24 and hydrogen bonding interactions involving pyridinic nitrogens and those hydrogens of methyl and aryl groups (see ESI Fig. S7†).
 | ||
| Fig. 2 (a) Molecular structure of 2 (top and side view, 50% probability level, H-atoms are omitted for clarity). (b) Illustration of the herringbone packing of 2 with alternating distances of the individual layers. | ||
The UV/vis absorption and emission spectra of DTPA and 1–3 in CH2Cl2 at room temperature are shown in Fig. 3a and their spectral data are summarized in Table 1. In contrast to the parent DTPA, which only exhibits a single absorption feature at 298 nm, 1–3 display two absorption bands with maxima in the range of 301–330 and 364–376 nm. The underlying electronic transitions are ascribed to π–π* transitions and ICT transitions from the central N-heterotriangulene as electron donor to the peripheral pyridyls as electron acceptors, respectively.14 Notable, the introduction of the first and the second 4-pyridyl unit into the parent N-heterotriangulene framework leads to significant bathochromic shifts. In contrast, the attachment of the third electron-withdrawing pyridyl has only marginal impact on the spectral features. Compounds 1–3 display strong emission with maxima between 439 and 448 nm. Interestingly, a pronounced positive solvatochromism is observed in the emission spectra when going from cyclohexane to methanol with shifts (Δλem) of 59 nm for 1, 63 nm for 2, and 74 nm for 3 (Fig. 3b, see also ESI Fig. S8–S10†).25 Protonation of the peripheral pyridyls with, for example, trifluoroacetic acid in CH2Cl2 leads to a strong bathochromic shift of both the absorption and the emission for 1–3 (see ESI Fig. S11 and S12†).26 This effect is strongest for 1, showing an absorption maximum shift (Δλabs) of 126 nm and an emission maximum shift (Δλem) of 182 nm compared to those of 2 (Δλabs 109 nm, Δλem 159 nm) and 3 (Δλabs 81 nm, Δλem 140 nm).
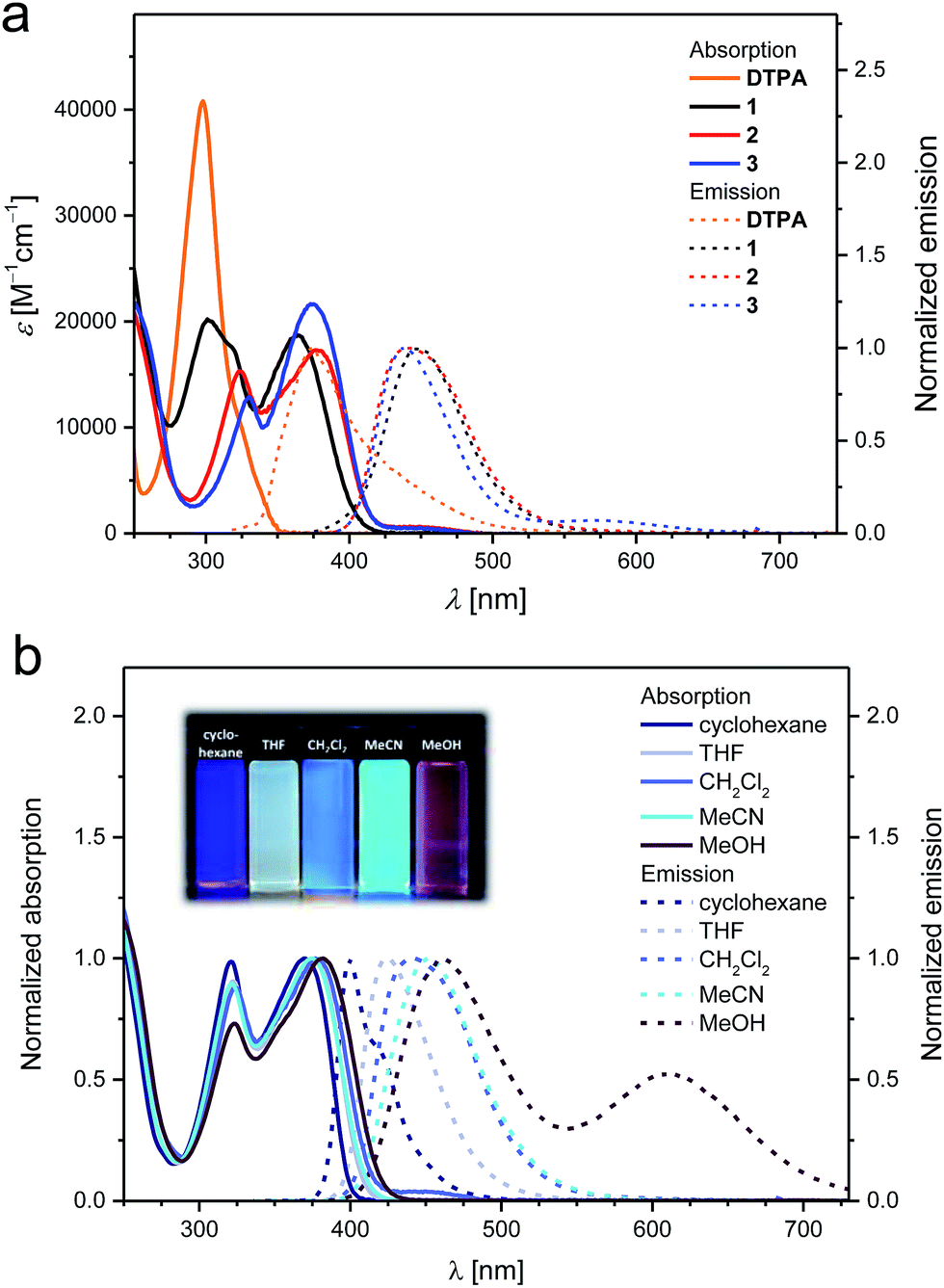 | ||
| Fig. 3 (a) UV/vis absorption (solid lines) and emission spectra (dashed lines) of DTPA, mono-1, bis-2, and trispyridyl substituted N-heterotriangulene 3 in CH2Cl2 at room temperature (DTPA: λexc = 298 nm, 1: λexc = 335 nm, 2: λexc = 339 nm, 3: λexc = 340 nm). (b) Exemplary normalized absorption (solid lines) and emission spectra (dashed lines) spectra of 2 measured in different solvents (cyclohexane: λexc = 315 nm, THF: λexc = 318 nm, CH2Cl2: λexc = 335 nm, MeCN: λexc = 313 nm, MeOH: λexc = 363 nm). | ||
| Dye | λ abs a [nm], (ε [M−1 cm−1]) | λ em a [nm] | Stokes shifta [cm−1] | E ox1/2 b [V] | HOMOc, (theo.)d [eV] | LUMOe, (theo.)d [eV] |
|
|
FFTiO2, FFZnOf | η TiO2, ηZnOf [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| a In CH2Cl2. b Half-wave potentials for oxidation (Eox1/2) recorded by cyclic voltammetry in CH2Cl2 with 0.1 M nBu4NPF6 (scan rate 150 mV s−1, referenced vs. Fc/Fc+). c HOMO level estimated from EHOMO = −(Eox1/2 + 4.8 eV).27 d Orbital energies calculated at the OLYP/6-311+G(d,p)//ωB97XD/6-31G(d) level of theory. e LUMO level estimated from ELUMO = EHOMO + Eg; optical band gap Eg corresponds to Eg = 1240λmax−1. f Photocurrent–voltage characteristics measured under simulated solar light conditions (1 sun and AM 1.5 conditions). | ||||||||||
| DTPA | 298, (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800) 800) |
374 | 6819 | +0.36 | −5.16, (−4.21) | −1.53, (−1.14) | — | — | — | — |
| 1 | 364, (18700) |
448 | 5151 | +0.42 | −5.22, (−4.38) | −2.16, (−1.86) | 0.76 | 0.63 | 0.54 | 0.26 |
| 2 | 376, (17300) |
443 | 4022 | +0.50 | −5.30, (−4.53) | −2.31, (−2.05) | 2.14, 1.31 | 0.70, 0.52 | 0.66, 0.56 | 0.98, 0.39 |
| 3 | 373, (21700) |
439 | 4031 | +0.56 | −5.36, (−4.66) | −2.36, (−2.16) | 2.34 | 0.70 | 0.45 | 0.75 |


Cyclic voltammetry studies of DTPA and 1–3 in CH2Cl2 (with 0.1 M nBu4NPF6, scan rate 150 mV s−1, vs. Fc/Fc+) at room temperature reveal reversible one-electron oxidations in all cases. In accordance with the electron-withdrawing nature of the pyridyl substituents the oxidation potential (Eox1/2) increases as a function of the number of attached pyridyl units from unsubstituted DTPA (+0.36 V) to 1 (+0.42 V), to 2 (+0.50 V), and to 3 (+0.56 V). This trend is nicely corroborated in density functional theory (DFT) calculations, that is, the calculated highest-occupied molecular orbital (HOMO) and lowest-unoccupied molecular orbital (LUMO) energies. They are all stabilized upon increasing the number of the pyridyl groups. The calculated Kohn–Sham frontier molecular orbital energies correlate with those estimated from the experimental data (see Table 1 and ESI Fig. S18†). In fact, the LUMO energies experience a more pronounced stabilization upon increasing the number of the electron-withdrawing pyridyls, when compared to the HOMO energies. It is worth mentioning that in all cases the LUMO energy is well above the conduction band of TiO2 (−4.3 eV) and ZnO (−4.7 eV) ensuring a sufficient driving force for electron injection.28
Considering all of the above-mentioned in concert, we investigated 1–3 as sensitizers for n-type DSSCs based on TiO2- and ZnO-based electrodes (for details regarding device fabrication and characterization, see ESI†). Interestingly, several groups have recently employed pyridyls as anchors for TiO2-based DSSCs,13–20 but, to the best of our knowledge, there are no examples in ZnO-based DSSCs.2b,13 This might relate to a number of intrinsic drawbacks associated with this type of linker. On one hand, it is the slow adsorption onto the electrode surface, leading to a low dye loading compared to their carboxylic acid congeners, and, on the other hand, the rather moderate electron injection rate.13 As a matter of fact, the latter factor is impacted by the number of pyridyls attached to the dye and the type of electrode.
As a first insight, the adsorption kinetics of 1–3 onto transparent TiO2 electrodes were monitored by means of absorption spectroscopy. Time-dependent adsorption assays reveal the growth of the absorption around 375 nm, which is similar to the spectral features observed for 1–3 in ethanol (see ESI Fig. S13†). Nevertheless, the absorption spectra of 1–3 on TiO2 electrodes indicate a slight red-shift and a broadening compared to the data in solution (see ESI Fig. S13†). From the latter we conclude good electronic communication between the electrode and 1–3.3,13,14 A direct comparison of the adsorption kinetics prompts to the fact that the number of pyridyl groups only slightly affects the time required for a complete electrode surface coverage, that is, ca. 30 h (see ESI Fig. S14†). As expected, the device performance relates to the adsorption time (Fig. 4). In particular, the fill factor (FF) gradually increases regardless of the number of pyridyl groups, reaching values of around 0.50 for 1 and 0.70 for 2 and 3. The low FFs noted for devices with 1 are quite likely due to the poor surface coverage, which is indicative for efficient electron recombination processes from the TiO2 electrode to the electrolyte. As a consequence, the electron density in the electrode is reduced, especially for devices with 1, and, in turn, the quasi-Fermi level of the sensitized electrode is lowered. In line with this argument is the fact that the lowest open-circuit voltage (Voc) in this series is noted for devices with 1 (Fig. 4). Our results imply that the binding of only one pyridyl group onto the electrode surface is inefficient, whereas the use of two or three pyridyl groups leads to enhanced device performances. Not surprisingly, the short-circuit current density (Jsc) is the lowest for devices with 1, while devices with 2 feature the highest Jsc after 30 h adsorption time followed by a gradual decrease. The latter Jsc and subsequently the efficiency values amount to approximately one fourth of the highest reported efficiencies of pyridyl-based DSSCs up to date.14–20 Finally, devices with 3 show decreasing Jsc with the adsorption time. This finding is likely to be due to aggregation on the electrode surface, as already shown in similar systems.3,13–15
 | ||
| Fig. 4 Figures-of-merit of DSSCs based on TiO2 electrodes with 1–3 (solid lines) and ZnO electrodes with 2 (dashed lines) at different adsorption times. | ||
In the next step, the incident-photon-to-current-efficiency (IPCE) spectra of the different devices were analyzed. Particular emphasis is placed on confirming the aforementioned trends for Jsc as a means to highlight the role of 1–3 as sensitizers (see Fig. 5). In detail, the IPCE spectra show a rising band at around 400 nm as a function of adsorption time. Maximum values are 23.1%, 58.4%, and 62.3% for devices with 1, 2, and 3, respectively (Fig. 5, see also ESI Fig. S15 and S16†). Overall, our data indicate that devices containing 2 with an efficiency (η) of 0.98% outperform those with 1 or 3.
 | ||
| Fig. 5 IPCE spectra of TiO2-based DSSCs with 2 at different adsorption times. | ||
To clarify the observed differences between the devices containing 2 and 3, the stability of them adsorbed onto TiO2-based electrodes was studied under constant irradiation (AM 1.5 and 1 sun) and thermal treatment (from room temperature to 300 °C with 50 °C steps each for 30 minutes). From UV/vis spectroscopy we infer that both dyes are stable for at least 90 minutes under constant irradiation and heating of up to 200 °C. Following that substantial changes with respect to the shape of the absorption spectra occurred (Fig. S18†). It is unlikely that a lack of dye stability is compromised and causes the observed device differences. Instead, one of the three pyridyl groups in 3, which remains unshielded from the electrolyte, is the inception for possible interactions with the redox electrolyte. To follow up on this notion, UV/vis absorption and emission spectroscopic assays were performed for 1–3 both in solution and with sensitized TiO2-based electrodes. To this end, a strong emission quenching was observed in ethanol solution upon electrolyte addition (Fig. S19†), suggesting a fast deactivation pathway through the population of the triplet excited state due to heavy atom effects. Simultaneously, a hypsochromic shift of the lowest-energy band sets in and a new band develops at around 300 nm (Fig. S20†). No shifts were, however, observed when the same experiments were performed with sensitized TiO2-based electrodes (Fig. S13 and S18†). Moreover, the IPCE data for 1–3 are the same and match perfectly the absorption features of the sensitized TiO2-based electrodes. It is reasonable to conclude that in a device under working conditions no significant interactions between the pyridyl linkers and the electrolyte takes place. Taking the aforementioned into concert, the most likely rationale for the differences between the devices containing 2 and 3 is the adsorption motif and/or arrangement at the electrode surface due to the unbound pyridyls.
Finally, 2 was used as probe to shed light onto the impact of the electrode material on the device performance. To this end, devices with ZnO-based electrodes featuring similar nanoparticle size and thickness as those used for the abovementioned experiments with TiO2-based DSSCs were fabricated (for experimental details, see ESI†). In this context, the basicity of ZnO films and the weak Zn–pyridine bond resulted in sluggish adsorption kinetics when compared to TiO2-based electrodes (Fig. 4).29 As such, the figures-of-merits for ZnO-based devices continuously grow until an adsorption time of 200 h is reached. Here, a maximum overall efficiency of 0.39% is realized (Fig. 4). Longer immersion times lead to a reduction of FF and Jsc, which might be ascribed to aggregation rather than to the intrinsic electrode instability.2c,30
Conclusions
In conclusion, we have synthesized a new family of fluorescent organic electron donor–acceptor dyes composed of a central N-heterotriangulene as electron donor decorated with 4-pyridyls as electron-withdrawing anchors and incorporated them as sensitizers in n-type DSSCs featuring TiO2 and ZnO electrodes. A combination of spectroscopic, electrochemical, and theoretical investigations assisted in documenting that as the number of pyridyl units is increased the HOMO as well as LUMO energies decreased. From absorption and emission assays we concluded pronounced solvatochromism and significant bathochromic shifts upon protonation. The latter feature bears great potential for fluorescent pH sensors. Moreover, we demonstrated that the pyridyl anchors in these novel electron donor–acceptor dyes enable, on one hand, the adsorption onto TiO2- and ZnO-based electrodes and, on the other hand, the effective electron injection upon light irradiation. In this context, the best device performances were realized with the dye that bears two pyridyl anchors. The latter seems to provide a good balance between efficient binding to the metal oxide surface and suppressing self-aggregation once immobilized onto the electrode surface. We are confident about future DSSC performance improvement by introduction of additional π-conjugated spacers, such as thiophenes, between the N-heterotriangulene core and the pyridyl anchors. This is primarily meant to enhance the absorption properties and, in turn, the light harvesting/photon collection characteristics.Acknowledgements
This work was supported by the “Solar Technologies Go Hybrid” (SolTech) initiative of the Free State of Bavaria. The German Research Foundation (DFG) through the Collaborative Research Center SFB 953 “Synthetic Carbon Allotropes”, the Cluster of Excellence “Engineering of Advanced Materials” (EAM) at the Friedrich-Alexander University (FAU) Erlangen-Nürnberg, and the Graduate School Molecular Science (GSMS) of FAU Erlangen-Nürnberg are gratefully acknowledged for their generous support.Notes and references
- (a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef; (b) M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- (a) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed; (b) J. A. Anta, E. Guillén and R. Tena-Zaera, J. Phys. Chem. C, 2012, 116, 11413–11425 CrossRef CAS; (c) R. D. Costa, F. Lodermeyer, R. Casillas and D. M. Guldi, Energy Environ. Sci., 2014, 7, 1281–1296 RSC; (d) F. Bella, C. Gerbaldi, C. Barolo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC; (e) M. Ye, X. Wen, M. Wang, J. Iocozzia, N. Zhang, C. Lin and Z. Lin, Mater. Today, 2015, 18, 155–162 CrossRef CAS.
- (a) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed; (b) M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed; (c) C.-P. Lee, R. Y.-Y. Lin, L.-Y. Lin, C.-T. Li, T.-C. Chu, S.-S. Sun, J. T. Lin and K.-C. Ho, RSC Adv., 2015, 5, 23810–23825 RSC; (d) J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed; (e) M.-E. Ragoussi and T. Torres, Chem. Commun., 2015, 51, 3957–3972 RSC.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- (a) L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–815 CrossRef CAS; (b) S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Péchy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202–1205 CrossRef CAS; (c) D. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923–1927 CrossRef CAS PubMed; (d) S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Péchy and M. Grätzel, Chem. Commun., 2008, 5194–5196 RSC.
- (a) S. Wang, J. Guo, L. He, H. Wang, J. Zhao and C. Lu, Synth. Met., 2013, 168, 1–8 CrossRef CAS; (b) C. A. Richard, Z. Pan, H.-Y. Hsu, S. Cekli, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2014, 6, 5221–5227 CrossRef CAS PubMed.
- K. Guo, K. Yan, X. Lu, Y. Qiu, Z. Liu, J. Sun, F. Yan, W. Guo and S. Yang, Org. Lett., 2012, 14, 2214–2217 CrossRef CAS PubMed.
- (a) S. Franco, J. Garín, N. M. de Baroja, R. Pérez-Tejada, J. Orduna, Y. Yu and M. Lira-Cantú, Org. Lett., 2012, 14, 752–755 CrossRef CAS PubMed; (b) R. Pérez-Tejada, N. M. de Baroja, S. Franco, L. Pellejà, J. Orduna, R. Andreu and J. Garín, Dyes Pigm., 2015, 123, 293–303 CrossRef.
- (a) Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 RSC; (b) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- (a) W.-H. Liu, I.-C. Wu, C.-H. Lai, C.-H. Lai, P.-T. Chou, Y.-T. Li, C.-L. Chen, Y.-Y. Hsu and Y. Chi, Chem. Commun., 2008, 5152–5154 RSC; (b) J.-H. Yum, D. P. Hagberg, S.-J. Moon, K. M. Karlsson, T. Marinado, L. Sun, A. Hagfeldt, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2009, 48, 1576–1580 CrossRef CAS PubMed.
- (a) D. Hellwinkel and M. Melan, Chem. Ber., 1971, 104, 1001–1016 CrossRef CAS; (b) J. E. Field and D. Venkataraman, Chem. Mater., 2002, 14, 962–964 CrossRef CAS.
- (a) D. Kim, C. Kim, H. Choi, K. Song, M.-S. Kang and J. Ko, J. Photochem. Photobiol., A, 2011, 219, 122–131 CrossRef CAS; (b) L. Cai, H. N. Tsao, W. Zhang, L. Wang, Z. Xue, M. Grätzel and B. Liu, Adv. Energy Mater., 2013, 3, 200–205 CrossRef CAS; (c) F. Wu, H. Liu, L. T. L. Lee, T. Chen, M. Wang and L. Zhu, Chin. J. Chem., 2015, 33, 925–933 CrossRef CAS; (d) F. Wu, L. T. L. Lee, J. Liu, S. Zhao, T. Chen, M. Wang, C. Zhong and L. Zhu, Synth. Met., 2015, 205, 70–77 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CAS.
- Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429–7433 CrossRef CAS PubMed.
- Y. Harima, T. Fujita, Y. Kano, I. Imae, K. Komaguchi, Y. Ooyama and J. Ohshita, J. Phys. Chem. C, 2013, 117, 16364–16370 CAS.
- (a) Y. Ooyama, T. Nagano, S. Inoue, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem.–Eur. J., 2011, 17, 14837–14843 CrossRef CAS PubMed; (b) Y. Ooyama, N. Yamaguchi, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. Commun., 2013, 49, 2548–2550 RSC; (c) Y. Ooyama, K. Uenaka, M. Kanda, T. Yamada, N. Shibayama and J. Ohshita, Dyes Pigm., 2015, 122, 40–45 CAS; (d) Y. Ooyama, K. Uenaka and J. Ohshita, Eur. J. Org. Chem., 2015, 3713–3720 CrossRef CAS; (e) Y. Harima, Y. Kano, T. Fujita, I. Imae, Y. Ooyama and J. Ohshita, RSC Adv., 2015, 5, 71387–71392 RSC.
- (a) D. Daphnomili, G. Landrou, S. P. Singh, A. Thomas, K. Yesudas, K. Bhanuprakash, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2012, 2, 12899–12908 RSC; (b) D. Daphnomili, G. D. Sharma, S. Biswas, K. R. J. Thomas and A. G. Coutsolelos, J. Photochem. Photobiol., A, 2013, 253, 88–96 CrossRef CAS; (c) J. Lu, X. Xu, Z. Li, K. Cao, J. Cui, Y. Zhang, Y. Shen, Y. Li, J. Zhu, S. Dai, W. Chen, Y. Cheng and M. Wang, Chem.–Asian J., 2013, 8, 956–962 CrossRef CAS PubMed; (d) C. Stangel, A. Bagaki, P. A. Angaridis, G. Charalambidis, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2014, 53, 11871–11881 CrossRef CAS PubMed.
- Y. Ooyama, Y. Hagiwara, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2479–2485 RSC.
- M. Cheng, X. Yang, J. Li, C. Chen, J. Zhao, Y. Wang and L. Sun, Chem.–Eur. J., 2012, 18, 16196–16202 CrossRef CAS PubMed.
- (a) B. Jin, W. Wu, X. Zhang, F. Guo, Q. Zhang and J. Hua, Chem. Lett., 2013, 42, 1271–1272 CrossRef CAS; (b) M.-D. Zhang, H.-X. Xie, X.-H. Ju, L. Qin, Q.-X. Yang, H.-G. Zheng and X.-F. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 634–641 RSC; (c) J. Mao, D. Wang, S.-H. Liu, Y. Hang, Y. Xu, Q. Zhang, W. Wu, P.-T. Chou and J. Hua, Asian J. Org. Chem., 2014, 3, 153–160 CrossRef CAS; (d) Y. Ooyama, K. Uenaka, T. Sato, N. Shibayama and J. Ohshita, RSC Adv., 2015, 5, 2531–2535 RSC.
- (a) S. Gottardi, K. Müller, J. C. Moreno-López, H. Yildrim, U. Meinhardt, M. Kivala, A. Kara and M. Stöhr, Adv. Mater. Interfaces, 2014, 1, 1300025 CrossRef; (b) A. T. Haedler, S. R. Beyer, N. Hammer, R. Hildner, M. Kivala, J. Köhler and H.-W. Schmidt, Chem.–Eur. J., 2014, 20, 11708–11718 CrossRef CAS PubMed; (c) N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119–1131 CrossRef CAS PubMed; (d) A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196–199 CrossRef CAS PubMed; (e) J.-C. Moreno-López, S. Gottardi, U. Meinhardt, H. Yildrim, A. Kara, M. Kivala and M. Stöhr, Chem.–Eur. J., 2016, 22, 581–589 CrossRef PubMed; (f) C. Steiner, B. D. Gliemann, U. Meinhardt, M. Gurrath, B. Meyer, M. Kivala and S. Maier, J. Phys. Chem. C, 2015, 119, 25945–25955 CrossRef CAS.
- Z. Fang, T.-L. Teo, L. Cai, Y.-H. Lai, A. Samoc and M. Samoc, Org. Lett., 2009, 11, 1–4 CrossRef CAS PubMed.
- F. Schlütter, F. Rossel, M. Kivala, V. Enkelmann, J.-P. Gisselbrecht, P. Ruffieux, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2013, 135, 4550–4557 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- P. Suppan, J. Photochem. Photobiol., A, 1990, 50, 293–330 CrossRef CAS.
- B. Hu, X. Chen, Y. Wang, P. Lu and Y. Wang, Chem.–Asian J., 2013, 8, 1144–1151 CrossRef CAS PubMed.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- Energy Efficiency and Renewable Energy Through Nanotechnology, ed. L. Zang, Springer, 2011 Search PubMed.
- (a) St. Hövel, C. Kolczewski, M. Wühn, J. Albers, K. Weiss, V. Staemmler and C. Wöll, J. Chem. Phys., 2000, 112, 3909–3916 CrossRef; (b) P. Joshi, V. Shewale, R. Pandey, V. Shanker, S. Hussain and S. P. Karna, Phys. Chem. Chem. Phys., 2011, 13, 476–479 RSC.
- J. Wu and D. Xue, Sci. Adv. Mater., 2011, 3, 127–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1441546. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14799b |
| This journal is © The Royal Society of Chemistry 2016 |