
Catalytic conversion of glucose into alkanediols over nickel-based catalysts: a mechanism study†
Abstract
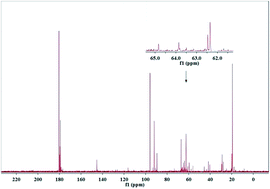
The conversion of isotope-labeled glucose (D-1-13C-glucose) into alkanediols was carried out in a batch reactor over a Ni–MgO–ZnO catalyst to reveal the C–C cleavage mechanisms. The unique role of the MgO–ZnO support was highlighted by 13C NMR and GC-MS analysis qualitatively and the MgO–ZnO favored isomerization of glucose to fructose. 13C NMR, GC-MS and HPLC analysis demonstrated that the C1 position of ethylene glycol, the C1 and C3 positions of 1,2-propanediol and the C1 position of glycerin were labeled with 13C, which is attributed to a C–C cleavage at D-1-13C-glucose's corresponding positions through retro-aldol condensation. A hydrogenolysis followed by hydrogenation pathway was proposed for glucose converted into alkanediols at 493 K with 6.0 MPa of H2 pressure over Ni based catalysts.
 Please wait while we load your content...
Please wait while we load your content...

