
Obtaining a high value branched bio-alkane from biomass-derived levulinic acid using RANEY® as hydrodeoxygenation catalyst†
 ab
ab
Abstract
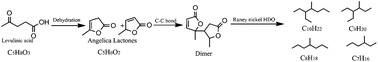
Compared to 5-HMF (C6H6O3), angelica lactone (C5H6O2) is a platform compound that has more potential for biomass-derived high performance bio-alkane fuel production due to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the molecular structure, leading to a C–C coupling intermediate (C10 self-aggregation dimer) and higher C : O ratio (2.5), resulting in lower hydrogen consumption for the subsequent hydrodeoxygenation process. Biomass-derived levulinic acid was used as the only starting raw material to produce C10 branched alkanes. First, carboxyl and carbonyl functional groups of levulinic acid under catalysis via intramolecular esterification and dehydration yielded angelica lactone, which included two isomers of angelica lactone (α-angelica lactone and β-angelica lactone). Secondly, angelica lactone di/trimers would be obtained by angelica lactone self-aggregation: α-angelica lactone and β-angelica lactone connecting via C–C bond coupling. Finally, these intermediate products are selectively hydrodeoxygenated over a RANEY® catalyst to obtain C7–C10 branched alkanes. Nearly a 90% yield can be achieved under 483 K and 5 MPa H2 and the C10 branched alkane product, 3-ethyl-4-methyl heptane, accounts for 75% of the same.
C bond in the molecular structure, leading to a C–C coupling intermediate (C10 self-aggregation dimer) and higher C : O ratio (2.5), resulting in lower hydrogen consumption for the subsequent hydrodeoxygenation process. Biomass-derived levulinic acid was used as the only starting raw material to produce C10 branched alkanes. First, carboxyl and carbonyl functional groups of levulinic acid under catalysis via intramolecular esterification and dehydration yielded angelica lactone, which included two isomers of angelica lactone (α-angelica lactone and β-angelica lactone). Secondly, angelica lactone di/trimers would be obtained by angelica lactone self-aggregation: α-angelica lactone and β-angelica lactone connecting via C–C bond coupling. Finally, these intermediate products are selectively hydrodeoxygenated over a RANEY® catalyst to obtain C7–C10 branched alkanes. Nearly a 90% yield can be achieved under 483 K and 5 MPa H2 and the C10 branched alkane product, 3-ethyl-4-methyl heptane, accounts for 75% of the same.
 Please wait while we load your content...
Please wait while we load your content...

