
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of 2,5-furandicarboxylic acid-based semi-aromatic polyamides: enzymatic polymerization kinetics, effect of diamine chain length and thermal properties†
Yi
Jiang‡
ab,
Dina
Maniar‡
a,
Albert J. J.
Woortman
a and
Katja
Loos
*ab
aDepartment of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl; Tel: +31-50 363 6867
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 11th July 2016
Abstract
2,5-Furandicarboxylic acid (FDCA)-based semi-aromatic polyamides are novel biobased alternatives to petrol-based semi-aromatic polyamides (polyphthalamides), that have a broad commercial interest as engineering thermoplastics and high performance materials. In this study, a series of FDCA-based semi-aromatic polyamides is successfully produced via Novozym®435 (N435, an immobilized form of Candida antarctica lipase b (CALB))-catalyzed polycondensation of (potentially) biobased dimethyl 2,5-furandicarboxylate and aliphatic diamines differing in chain length (C4–C12), using a one-stage method at 90 °C in toluene. The obtained polyamides reach high weight-average molecular weights ranging from 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 to 48300 g mol−1; and N435 shows the highest selectivity towards 1,8-octanediame (C8). MALDI-ToF MS analysis indicates that no byproducts are formed during the enzymatic polymerization. Study of the kinetics of the enzymatic polymerization suggests that phase separation of FDCA-based oligoamides/polyamides takes place in the early stage of polymerization, and the isolated products undergo an enzyme-catalyzed solid-state polymerization. However, the isolation yields of the purified products from the enzymatic polymerizations are less than ∼50% due to the production of a large amount of low molecular weight products that are washed away during the purification steps. Furthermore, the thermal properties of the enzymatic FDCA-based semi-aromatic polyamides are carefully investigated, and compared to those of the FDCA-based and petrol-based counterparts produced via conventional synthesis techniques as reported in literature.
800 to 48300 g mol−1; and N435 shows the highest selectivity towards 1,8-octanediame (C8). MALDI-ToF MS analysis indicates that no byproducts are formed during the enzymatic polymerization. Study of the kinetics of the enzymatic polymerization suggests that phase separation of FDCA-based oligoamides/polyamides takes place in the early stage of polymerization, and the isolated products undergo an enzyme-catalyzed solid-state polymerization. However, the isolation yields of the purified products from the enzymatic polymerizations are less than ∼50% due to the production of a large amount of low molecular weight products that are washed away during the purification steps. Furthermore, the thermal properties of the enzymatic FDCA-based semi-aromatic polyamides are carefully investigated, and compared to those of the FDCA-based and petrol-based counterparts produced via conventional synthesis techniques as reported in literature.
Introduction
2,5-Furandicarboxylic acid (FDCA) can be produced by chemical or biocatalytic oxidation of 5-(hydroxymethyl)furfural (HMF) that is usually derived from various renewable carbohydrates.1–3 Currently, FDCA is already commercially available.4 It can be expected that the price of biobased FDCA will be comparable to or even cheaper than those of the biobased and petrol-based TPA.5,6 Therefore, FDCA, currently the most promising green alternative to TPA, has great potential for industrial applications such as in the synthesis of novel aromatic polymers. These polymers possess similar or even better properties than those of their petrol-based counterparts.2,7–9Among aromatic polymers, semi-aromatic polyamides (polyphthalamides) consist of both aliphatic and aromatic monomeric units which are linked by amide bonds in the main chain. These polymers are commonly used as engineering thermoplastics and high performance materials,10–12 owing to their good chemical/abrasion/corrosion resistance, excellent mechanical properties and many other appealing attributes. Semi-aromatic polyamides have found various applications in many fields, for example, in the automotive industry, electrical and electronics appliances, food contact materials, medical devices, photovoltaic panels and parts, and oil and gas polymers.
In general, semi-aromatic polyamides can be produced via step-growth polycondensation of aromatic diacid derivatives and aliphatic diamines at both laboratory and industrial scale,13 usually at elevated temperatures above 200 °C. However, such high temperatures may induce many undesirable side-reactions, for example, pyrolysis of aromatic diacids, N-methylation of (poly)amides, cross-linking of polymer chains, and gel formation, as well as, self-condensation and cyclization of diamines.8,13 These side-reactions result in not only the formation of low molecular weight products and discoloration, but property detriments of the obtained polymeric materials.
Fortunately, the side-reactions can be greatly suppressed by using enzymatic polymerizations, due to the mild reaction conditions and the high catalytic specificity of the biocatalysts. Enzymatic polymerizations are defined as the “in vitro (in the test tubes) chemical synthesis of polymers via a non-biosynthetic (non-metabolic) approach using an isolated enzyme as the catalyst”.14 It is an alternative and powerful pathway for the production of commodity polymers, which can compete with conventional chemical synthesis approaches and physical modifications.15–21 Enzymatic polymerizations are also capable of producing various novel polymers that are difficult to access via conventional approaches. Moreover, enzymatic polymerizations are clean processes, which can provide many advantageous sustainable aspects such as energy and material saving, non-toxic renewable enzyme catalysts, and gentle carbon footprint.22 Furthermore, by utilizing biobased monomers in enzymatic polymerizations, the energy and material consumption can be further reduced, and the generation of hazardous waste and emissions can be greatly minimized. This is essential for achieving a green polymer industry, and will eventually be beneficial for realizing and maintaining a sustainable society.
At present, 4 EC enzyme classes, including oxidoreductases (EC 1), transferases (EC 2), hydrolases (EC 3) and ligases (EC 6), are frequently used to catalyze or induce polymerizations.23 Polymer classes produced via enzymatic polymerizations include vinyl polymers,24,25 polysaccharides,26 polyesters,19,22 polyamides,27–29 and so on.21,23
In principle, enzymes that can catalyze the formation of amide bonds are suitable biocatalysts for the synthesis of polyamides.27,29 Currently, hydrolases such as proteases, esterases (especially lipases) and other enzymes, are commonly applied for the biocatalytic synthesis of polypeptides and synthetic polyamides. Among hydrolases, Candida antarctica lipase b (CALB), especially its immobilized formulation Novozym®435 (N435), is the primary enzyme catalyst used for enzymatic polyamide synthesis, as it possesses a broad substrate specificity, high selectivity, and excellent and stable catalytic activity. Many synthetic oligoamides and polyamides were successfully synthesized via CALB-catalyzed polymerization, for example: aliphatic oligoamides,30 semi-aromatic oligoamides,31 aliphatic polyamides (nylons),32–36 silicone aromatic polyamides,37 and poly(ester amide)s.34,38
Previously, we demonstrated that CALB possesses high catalytic activity towards rigid furan monomers including dimethyl 2,5-furandicarboxylate (DMFDCA) and 2,5-bis(hydroxymethyl)furan (BHMF),39,40 and the enzymatic polymerizations yielded various FDCA-based and BHMF-based semi-aromatic polyesters with high  ’s (weight-average molecular weights) of up to 100000 g mol−1. Recently we applied the well-established methodology from the enzymatic polymerization of biobased furan polyesters to prepare a FDCA-based semi-aromatic polyamide, poly(octamethylene furanamide) (PA8F), starting from DMFDCA and 1,8-octanediamine (1,8-ODA) and using N435 as the biocatalyst.41 High molecular weight PA8F was successfully produced via a one-stage method in toluene, with a high
’s (weight-average molecular weights) of up to 100000 g mol−1. Recently we applied the well-established methodology from the enzymatic polymerization of biobased furan polyesters to prepare a FDCA-based semi-aromatic polyamide, poly(octamethylene furanamide) (PA8F), starting from DMFDCA and 1,8-octanediamine (1,8-ODA) and using N435 as the biocatalyst.41 High molecular weight PA8F was successfully produced via a one-stage method in toluene, with a high  and
and  of 13400 and 48300 g mol−1, respectively. On the other hand, limited studies are available on the production of FDCA-based semi-aromatic polyamides by direct polycondensation of FDCA derivatives and aliphatic diamines using conventional synthesis techniques;42–46 and the resulting polyamides possessed relatively lower molecular weights (
of 13400 and 48300 g mol−1, respectively. On the other hand, limited studies are available on the production of FDCA-based semi-aromatic polyamides by direct polycondensation of FDCA derivatives and aliphatic diamines using conventional synthesis techniques;42–46 and the resulting polyamides possessed relatively lower molecular weights ( = 4300–10000 g mol−1) due to the decarboxylation of FDCA and extensive occurrence of N-methylation of (poly)amides at elevated temperatures.8,45–48 Therefore, enzymatic polymerizations are an appealing alternative approach for the production of FDCA-based semi-aromatic polyamides with high molecular weights. However, the isolation yields of the purified PA8F from the enzymatic polymerizations were less than ∼50% despite the different polymerization conditions employed, as the resulting polyamides possess a low solubility in the reaction media.
= 4300–10000 g mol−1) due to the decarboxylation of FDCA and extensive occurrence of N-methylation of (poly)amides at elevated temperatures.8,45–48 Therefore, enzymatic polymerizations are an appealing alternative approach for the production of FDCA-based semi-aromatic polyamides with high molecular weights. However, the isolation yields of the purified PA8F from the enzymatic polymerizations were less than ∼50% despite the different polymerization conditions employed, as the resulting polyamides possess a low solubility in the reaction media.
In this study, to better understand the one-stage enzymatic polymerization of FDCA-based semi-aromatic polyamides, the enzymatic polymerization kinetics is studied by NMR, SEC and MALDI-ToF MS, using DMFDCA and 1,8-ODA as model compounds. Moreover, we extend our research to synthesize a series of sustainable FDCA-based semi-aromatic polyamides by using various (potentially) biobased aliphatic diamines differing in chain length (see Scheme 1). In addition, we investigated the chemical structures, end groups, and thermal properties of the obtained polyamides. Furthermore, we compared the thermal properties of the enzymatic FDCA-based semi-aromatic polyamides to those of the FDCA-based42–44 and TPA-based counterparts49–52 produced via conventional synthesis approaches as reported in literature.
 | ||
| Scheme 1 Enzymatic synthesis of FDCA-based semi-aromatic polyamides (PAXF) via N435-catalyzed polycondensation of DMFDCA and aliphatic diamines at 90 °C in toluene. | ||
Experimental
Materials
N435 (≥5000 U g−1), 1,4-butanediamine (1,4-BDA, 99%), 1,6-hexanediamine (1,6-HDA, 98%), 1,8-octanediamine (1,8-ODA, 98%), 1,10-decanediamine (1,10-DDA, 97%), 1,12-dodecanediamine (1,12-DODA, 98%), toluene (anhydrous, 99.8%), formic acid (puriss, ≥98%), 1,4-dioxane (≥99%), molecular sieves (4 Å), dimethyl sulfoxide (DMSO, HPLC grade), dimethyl sulfoxide-d6 (DMSO-d6), trifluoroacetic acid-d1 (TFA-d1), and potassium trifluoroacetate (KTFA, 98%) were purchased from Sigma-Aldrich. DMFDCA (97%) was ordered from Fluorochem UK. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, ≥99%) was acquired from TCI Europe. Dithranol (≥98%) was purchased from Fluka. Methanol (99.8%) was purchased from Labscan.The diamines, including 1,4-BDA, 1,6-HDA, 1,8-ODA, 1,10-DDA, and 1,12-DODA, were purified by sublimation under reduced pressure and then stored in a desiccator. N435 was pre-dried as reported previously.41,53,54 The molecular sieves (4 Å) were pre-activated at 200 °C in vacuo. The other chemicals were used without further purification.
Procedure for N435-catalyzed polycondensation of DMFDCA and various aliphatic diamines via a one-stage method in toluene
Pre-dried N435 (20 wt%, in relation to the total amount of all monomers) and pre-activated 4 Å molecular sieves (200 wt%) were fed into a 25 mL round-bottom flask filled with nitrogen. Subsequently DMFDCA (0.5000 g, 2.715 mmol), an aliphatic diamine (0.1917–0.4358 g, 2.715 mmol), and anhydrous toluene (500 wt%) were introduced into the flask, thereafter the flask was sealed. The flask was then placed in an oil bath, where after the reaction mixture was magnetically stirred at 90 °C for 72 h.After the polymerization, toluene was evaporated by air-blowing at room temperature. Then the products were dissolved by formic acid (∼15 mL). After that, N435 and molecular sieves were removed by normal filtration using filter paper. Then the N435, molecular sieves and the used filter paper were washed three times with formic acid (∼10 mL). All the solutions obtained were combined and then concentrated by a rotary evaporator at 40 °C under reduced pressure (20–40 mbar). The concentrated solution was added dropwise into an excess amount of 1,4-dioxane. The crude products were collected by centrifugation (30 minutes, 4500 rpm, 12 °C) and decantation at room temperature. After that, the crude products obtained were dissolved again with formic acid (∼10 mL) and then added dropwise into excess of methanol. Then the methanol solution with the precipitates was stored at −20 °C for several hours. Subsequently, the precipitated products were collected by centrifugation (30 minutes, 4500 rpm, 0 °C) and then dried in vacuo at 40 °C for 3 days. Finally, the obtained FDCA-based semi-aromatic polyamides were stored in vacuo at room temperature prior to analysis.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, furan), 3.14 (4H, m, –NH–CH2–, from 1,4-BDA), 1.55 (4H, m, –NH–CH2–CH2–, from 1,4-BDA), 2.81 (t, –CH2–NH2, end groups from 1,4-BDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.16 (–CO–NH–, from DMFDCA), 148.14 (–NH–CO–C(O)CH–, furan), 114.26 (CH–, furan), 38.19 (–CO–NH–CH2–, from 1,4-BDA), 26.84 (–CO–NH–CH2–CH2–, from 1,4-BDA). Isolation yield: 0.0853 g, 7%.
CH–, furan), 3.24 (4H, m, –NH–CH2–, from 1,6-HDA), 1.51 (4H, m, –NH–CH2–CH2–, from 1,6-HDA), 1.31 (4H, m, –NH–CH2–CH2–CH2–, from 1,6-HDA), 3.84 (s, –OCH3, end groups from DMFDCA), 2.76 (t, –CH2–NH2, end groups from 1,6-HDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.33 (–CO–NH–, from DMFDCA), 148.22 (–NH–CO–C(O)CH–, furan), 114.44 (CH–, furan), 38.56 (–CO–NH–CH2–, from 1,6-HDA), 29.31 (–CO–NH–CH2–CH2–, from 1,6-HDA), 26.22 (–CO–NH–CH2–CH2–CH2–, from 1,6-HDA). Isolation yield: 0.2084 g, 23%.
CH–, furan), 3.22 (4H, m, –NH–CH2–, from 1,10-DDA), 1.47 (4H, m, –NH–CH2–CH2–, from 1,10-DDA), 1.23 (12H, m, –NH–CH2–CH2–CH2–CH2–CH2–, from 1,10-DDA), 3.83 (s, –OCH3, end groups from DMFDCA), 2.73 (t, –CH2–NH2, end groups from 1,10-DDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.07 (–CO–NH–, from DMFDCA), 148.14 (–NH–CO–C(O)CH–, furan), 114.18 (CH–, furan), 38.48 (–CO–NH–CH2–, from 1,10-DDA), 29.25(–CO–NH–CH2–CH2–, from 1,10-DDA), 28.91 (–CO–NH–CH2–CH2–CH2–CH2–CH2–, from 1,10-DDA), 28.73 (–CO–NH–CH2–CH2–CH2–CH2–, from 1,10-DDA), 26.43 (–CO–NH–CH2–CH2–CH2–, from 1,10-DDA). Isolation yield: 0.3150 g, 35%.
CH–, furan), 3.21 (4H, m, –NH–CH2–, from 1,12-DODA), 1.46 (4H, m, –NH–CH2–CH2–, from 1,12-DODA), 1.21 (16H, m, –NH–CH2–CH2–CH2–CH2–CH2–CH2–, from 1,12-DODA), 3.82 (s, –OCH3, end groups from DMFDCA); 13C NMR (75 MHz, TFA-d1, δ, ppm): 160.69 (–CO–NH–, from DMFDCA), 147.70 (–NH–CO–C(O)CH–, furan), 117.60 (CH–, furan), 41.26 (–CO–NH–CH2–, from 1,12-DODA), 29.40 (–CO–NH–CH2–CH2–CH2–CH2–CH2–CH2–, from 1,12-DODA), 29.06 (–CO–NH–CH2–CH2–, from 1,12-DODA), 28.82 (–CO–NH–CH2–CH2–CH2–CH2–, from 1,12-DODA), 26.71 (–CO–NH–CH2–CH2–CH2–, from 1,12-DODA). Isolation yield: 0.1473 g, 16%.
C–H stretching vibrations of the furan ring); 2921–2932, 2851–2865 (asymmetric and symmetric C–H stretching vibrations); 1625–1646 (CO stretching vibrations); 1570–1573 (aromatic CC bending vibrations); 1524–1531 (N–H bending vibrations); 1456–1472, 1435–1438 (C–H deformation and wagging vibrations); 1365–1384 (C–H rocking vibrations); 1279–1292 (C–N stretching vibrations); 1160–1166, 1012–1016 (C–O–C ring vibrations, furan ring); 961–968, 819–820, 757 (C–H out-of-plane deformation vibrations, furan ring); 718 (–(CH2)n–, rocking vibrations); 672 (C–H bending vibrations); 646–650 (N–H wagging vibrations).
CH–, furan), 3.14 (4H, m, –NH–CH2–, from 1,4-BDA), 1.55 (4H, m, –NH–CH2–CH2–, from 1,4-BDA), 2.81 (t, –CH2–NH2, end groups from 1,4-BDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.16 (–CO–NH–, from DMFDCA), 148.14 (–NH–CO–C(O)CH–, furan), 114.26 (CH–, furan), 38.19 (–CO–NH–CH2–, from 1,4-BDA), 26.84 (–CO–NH–CH2–CH2–, from 1,4-BDA). Isolation yield: 0.0853 g, 7%.
CH–, furan), 3.24 (4H, m, –NH–CH2–, from 1,6-HDA), 1.51 (4H, m, –NH–CH2–CH2–, from 1,6-HDA), 1.31 (4H, m, –NH–CH2–CH2–CH2–, from 1,6-HDA), 3.84 (s, –OCH3, end groups from DMFDCA), 2.76 (t, –CH2–NH2, end groups from 1,6-HDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.33 (–CO–NH–, from DMFDCA), 148.22 (–NH–CO–C(O)CH–, furan), 114.44 (CH–, furan), 38.56 (–CO–NH–CH2–, from 1,6-HDA), 29.31 (–CO–NH–CH2–CH2–, from 1,6-HDA), 26.22 (–CO–NH–CH2–CH2–CH2–, from 1,6-HDA). Isolation yield: 0.2084 g, 23%.
CH–, furan), 3.22 (4H, m, –NH–CH2–, from 1,10-DDA), 1.47 (4H, m, –NH–CH2–CH2–, from 1,10-DDA), 1.23 (12H, m, –NH–CH2–CH2–CH2–CH2–CH2–, from 1,10-DDA), 3.83 (s, –OCH3, end groups from DMFDCA), 2.73 (t, –CH2–NH2, end groups from 1,10-DDA); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 157.07 (–CO–NH–, from DMFDCA), 148.14 (–NH–CO–C(O)CH–, furan), 114.18 (CH–, furan), 38.48 (–CO–NH–CH2–, from 1,10-DDA), 29.25(–CO–NH–CH2–CH2–, from 1,10-DDA), 28.91 (–CO–NH–CH2–CH2–CH2–CH2–CH2–, from 1,10-DDA), 28.73 (–CO–NH–CH2–CH2–CH2–CH2–, from 1,10-DDA), 26.43 (–CO–NH–CH2–CH2–CH2–, from 1,10-DDA). Isolation yield: 0.3150 g, 35%.
CH–, furan), 3.21 (4H, m, –NH–CH2–, from 1,12-DODA), 1.46 (4H, m, –NH–CH2–CH2–, from 1,12-DODA), 1.21 (16H, m, –NH–CH2–CH2–CH2–CH2–CH2–CH2–, from 1,12-DODA), 3.82 (s, –OCH3, end groups from DMFDCA); 13C NMR (75 MHz, TFA-d1, δ, ppm): 160.69 (–CO–NH–, from DMFDCA), 147.70 (–NH–CO–C(O)CH–, furan), 117.60 (CH–, furan), 41.26 (–CO–NH–CH2–, from 1,12-DODA), 29.40 (–CO–NH–CH2–CH2–CH2–CH2–CH2–CH2–, from 1,12-DODA), 29.06 (–CO–NH–CH2–CH2–, from 1,12-DODA), 28.82 (–CO–NH–CH2–CH2–CH2–CH2–, from 1,12-DODA), 26.71 (–CO–NH–CH2–CH2–CH2–, from 1,12-DODA). Isolation yield: 0.1473 g, 16%.
C–H stretching vibrations of the furan ring); 2921–2932, 2851–2865 (asymmetric and symmetric C–H stretching vibrations); 1625–1646 (CO stretching vibrations); 1570–1573 (aromatic CC bending vibrations); 1524–1531 (N–H bending vibrations); 1456–1472, 1435–1438 (C–H deformation and wagging vibrations); 1365–1384 (C–H rocking vibrations); 1279–1292 (C–N stretching vibrations); 1160–1166, 1012–1016 (C–O–C ring vibrations, furan ring); 961–968, 819–820, 757 (C–H out-of-plane deformation vibrations, furan ring); 718 (–(CH2)n–, rocking vibrations); 672 (C–H bending vibrations); 646–650 (N–H wagging vibrations).
Enzymatic polymerization kinetics study: the N435-catalyzed polycondensation of DMFDCA and 1,8-ODA via the one-stage method in toluene
Instrumental methods
1H NMR spectra were recorded on a 400 MHz Varian VXR spectrometer and 13C NMR spectra were recorded on a 300 MHz Varian VXR spectrometer. The solvent was DMSO-d6 or TFA-d1. The reported chemical shifts were referenced to the resonances of the residual solvent or tetramethylsilane (TMS). The number-average molecular weight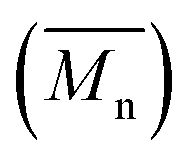 was determined by 1H NMR according to literature.39–41
was determined by 1H NMR according to literature.39–41
Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra were recorded on a Bruker IFS88 FT-IR spectrometer, with 128 scans for each sample.
The molecular weights of the crude PA8F from the enzymatic polymerization kinetics study were measured at 80 °C using an Agilent size exclusion chromatography (SEC) system (Agilent Technologies 1260 Infinity) from PSS (Mainz, Germany). The SEC system was equipped with three detectors (an Agilent refractive index detector G1362A 1260 RID, a PSS viscometer detector ETA-2010, and a PSS MALLS detector SLD 7000), and four columns (a PFG guard-column and three PFG SEC columns 100, 300 and 4000 Å). The detectors were kept at 45 °C, 60 °C and room temperature, respectively. The eluent was DMSO (HPLC grade) with LiBr (0.05 M), with a flow rate of 0.5 mL min−1.  and
and  were determined by conventional calibration using a calibration curve generated by pullulan standards (from PSS,
were determined by conventional calibration using a calibration curve generated by pullulan standards (from PSS,  = 342 to 805000 g mol−1).
= 342 to 805000 g mol−1).
The molecular weights of the purified PA4F, PA6F, PA10F and PA12F were determined by SEC on a Viscotec GPCmax system equipped with model 302 TDA detectors, a guard column (PSS-GRAM, 10 μm, 5 cm) and two analytical columns (PSS-GRAM-1000/30 Å, 10 μm, 30 cm). The eluent was DMF (HPLC grade) with LiBr (0.01 M), with a flow rate of 1 mL min−1. 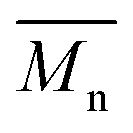 and
and  were calculated by conventional calibration, using a calibration curve generated by polymethylmethacrylate (PMMA) standards (from PSS,
were calculated by conventional calibration, using a calibration curve generated by polymethylmethacrylate (PMMA) standards (from PSS, 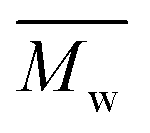 = 2460–655000 g mol−1).
= 2460–655000 g mol−1).
Thermal transitions of the synthetic FDCA-based semi-aromatic polyamides were characterized by a TA-Instruments Q1000 DSC (differential scanning calorimetry), with a heating and cooling rate of 10 °C min−1. Before the standard DSC measurement, the tested polyamides were heated up to 100 °C at 10 °C min−1, kept at this temperature for 5 min, and then cooled down to room temperature, to remove the remaining solvents and water.
Thermal stability measurements of the obtained FDCA-based semi-aromatic polyamides were performed on a Perkin-Elmer thermogravimetric analyzer TGA7 under nitrogen environment, with a scan rate of 10 °C min−1. Before the standard thermal gravimetric analysis (TGA), the tested polyamides were heated up to 100 °C and then kept at this temperature for 0.5 h, to remove the remaining solvents and water.
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-ToF MS) measurements were performed on a Biosystems Voyager-DE PRO spectrometer in positive and linear mode. The used matrix, solvent and cationization agent were dithranol, HFIP and KTFA, respectively. At first, dithranol (20 mg mL−1), KTFA (5 mg mL−1) and a polymer sample (1–2 mg mL−1) were premixed in a ratio of 5:1:5. Then the mixture was hand-spotted on a stainless steel plate and left to dry afterwards. Polyamide species having different end groups were determined by the following equation:
| MP = MEG + (n × MRU) + MK+ |
Results and discussion
Enzymatic polymerization kinetics study
In our previous study, we found that the one-stage enzymatic polycondensation of DMFDCA and 1,8-ODA in toluene resulted in PA8F with high molecular weights but a low isolation yield (<∼50%, purified products).41 To better understand the one-stage enzymatic polymerization in toluene, the enzymatic polymerization kinetics was investigated by 1H NMR, SEC and MALDI-ToF MS (see Fig. 1–5, and Tables S1 and S2 in the ESI†). | ||
| Fig. 1 1H NMR spectra of the solution mixture from the N435-catalyzed polycondensation of DMFDCA and 1,8-ODA at 90 °C in toluene. | ||
 | ||
| Fig. 2 1H NMR spectra of the solution mixture from the control reaction of DMFDCA and 1,8-ODA at 90 °C in toluene in the absence of N435. | ||
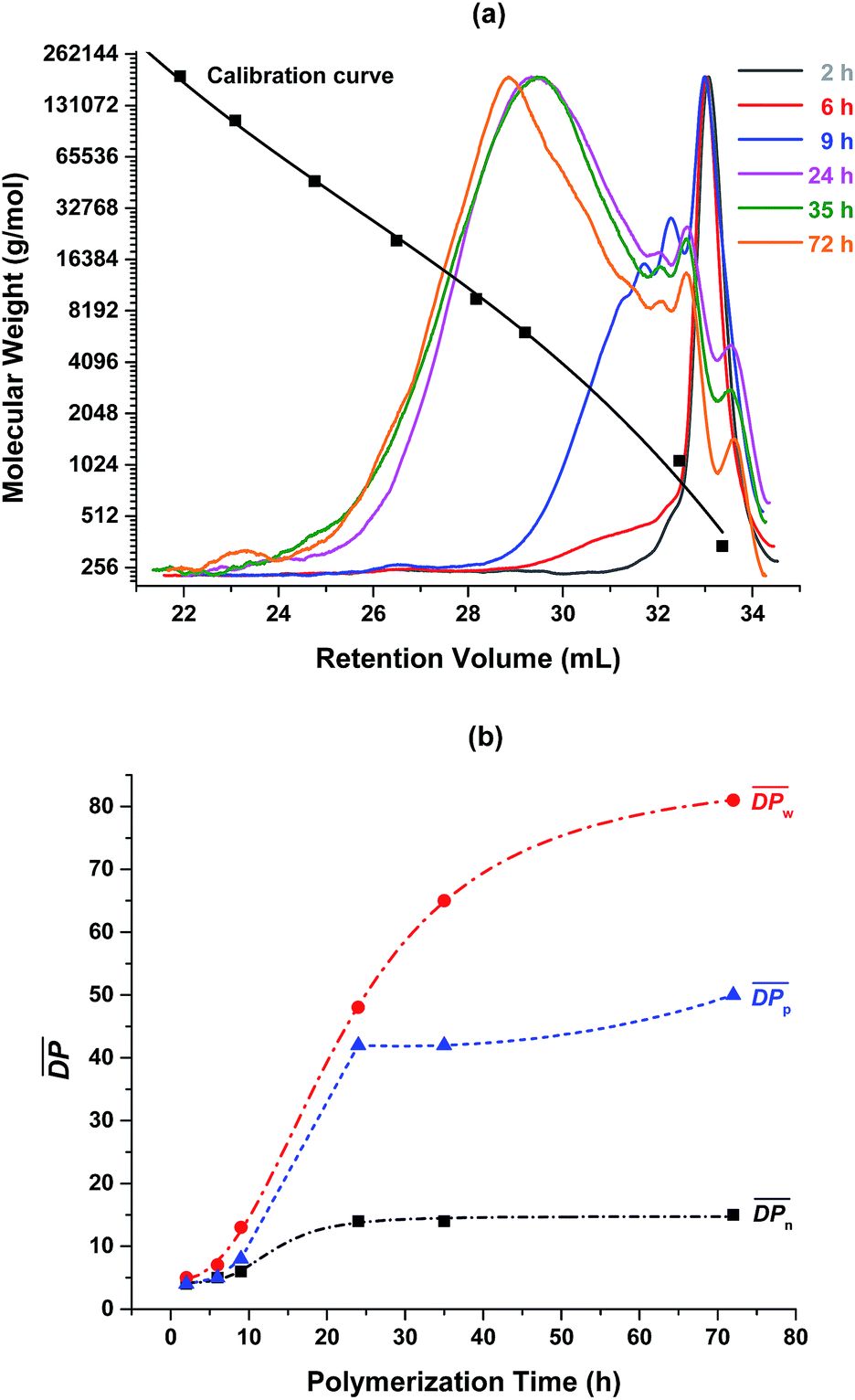 | ||
Fig. 3 (a) SEC elution curves of the obtained crude PA8F from the N435-catalyzed polycondensation of DMFDCA and 1,8-ODA in toluene at different polymerization times; and (b)  , ,  and and  of the crude PA8F determined by SEC as a function of the polymerization time. Herein, of the crude PA8F determined by SEC as a function of the polymerization time. Herein,  , , 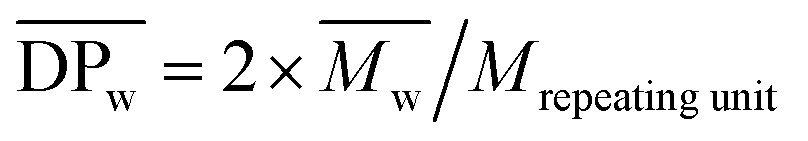 , ,  , where Mrepeating unit is the molecular mass of the repeating units. , where Mrepeating unit is the molecular mass of the repeating units. | ||
 | ||
Fig. 4 Evolution of the cumulative weight fractions of the obtained crude PA8F with different degrees of polymerization ( ) as a function of polymerization time. The crude PA8F was obtained from the enzymatic kinetics study; and ) as a function of polymerization time. The crude PA8F was obtained from the enzymatic kinetics study; and  and cumulative weight fractions were determined by SEC. and cumulative weight fractions were determined by SEC. | ||
 | ||
| Fig. 5 MALDI-ToF MS spectra of the obtained crude PA8F from the enzymatic kinetics study at different polymerization times. | ||
We noticed that FDCA-based semi-aromatic oligoamides were already produced within 30 minutes of reaction time, as proven by the appearance of a new signal at 3.83 ppm that is ascribed to the methoxyl groups of oligoamides (see Fig. 1). Moreover, the proton signal assigned to the methoxyl groups of DMFDCA disappeared completely after 2 h oligomerization, indicating that all DMFDCA monomer was converted to oligoamides. This agreed well with our previous result, which proved that 2 h oligomerization is sufficient for the enzymatic polycondensation of aliphatic polyesters.54
However, when the polymerization time was extended from 3 to 9 h, the relative intensity of the peak ascribed to the methoxyl groups of the resulting oligoamides/polyamides decreased significantly. This is due to the precipitation of the resulting products because of their low solubility. Moreover, no resonances can be assigned to the protons of the FDCA-based oligoamides/polyamides after ∼22 h reaction. This suggested that no resulting products were identified in the reaction mixture, as they were totally isolated from the reaction media due to the precipitation.
In contrast to this, the monomer DMFDCA and the resulting FDCA-based oligoamides/polyamides were clearly detected from the control reaction in the absence of N435 after 0.5–71 h reaction (see Fig. 2). Meanwhile, more oligoamides/polyamides were prepared at a longer polymerization time, as the relative intensity of the peak ascribed to the monomer DMFDCA decreases gradually with the increase of polymerization time. This indicated that the condensation between DMFDCA and 1,8-ODA can occur in the absence of catalysts, but the reaction rate is rather low. In other words, from Fig. 1 and 2 we can draw the conclusion that the polycondensation is indeed catalyzed by N435, which resulted in high molecular weight PA8F as reported in our previous study.41
The enzymatic polymerization kinetics was also investigated by SEC (Fig. 3 and 4 and Tables S1 and S2†). Fig. 3 depicts the SEC elution curves of the crude PA8F from the enzymatic kinetics study and a plot of degrees of polymerization  as a function of the polymerization time. When the polymerization time was extended from 2 to 24 h, the major retention volume of the obtained crude PA8F shifted significantly to a lower value (Fig. 3a), indicating the formation of higher molecular weight products at a longer polymerization time. As shown in Fig. 3b and Table S1,† the corresponding number-average degree of polymerization
as a function of the polymerization time. When the polymerization time was extended from 2 to 24 h, the major retention volume of the obtained crude PA8F shifted significantly to a lower value (Fig. 3a), indicating the formation of higher molecular weight products at a longer polymerization time. As shown in Fig. 3b and Table S1,† the corresponding number-average degree of polymerization  , weight-average degree of polymerization
, weight-average degree of polymerization  , and peak degree of polymerization (
, and peak degree of polymerization ( , the major retention volume) increased significantly, from 4, 5 and 4 to 14, 48 and 42, respectively. Upon further increasing the polymerization time from 24 to 72 h, the
, the major retention volume) increased significantly, from 4, 5 and 4 to 14, 48 and 42, respectively. Upon further increasing the polymerization time from 24 to 72 h, the  and
and  slightly increased, from 14 and 42 to 15 and 50, respectively. On the contrary, the
slightly increased, from 14 and 42 to 15 and 50, respectively. On the contrary, the  showed a significant increase from 48 to 81, even though all the resulting PA8F was phase separated from the reaction after ∼22 h reaction as indicated by the NMR study discussed above. This is consistent with the MALDI-ToF MS results which also confirmed that the enzymatic polymerization at longer reaction times resulted in longer chain PA8F (Fig. 5). The chain growth of the isolated products after ∼22 h reaction could be explained by the N435-catalyzed solid-state polymerization, as postulated in our previous study.41
showed a significant increase from 48 to 81, even though all the resulting PA8F was phase separated from the reaction after ∼22 h reaction as indicated by the NMR study discussed above. This is consistent with the MALDI-ToF MS results which also confirmed that the enzymatic polymerization at longer reaction times resulted in longer chain PA8F (Fig. 5). The chain growth of the isolated products after ∼22 h reaction could be explained by the N435-catalyzed solid-state polymerization, as postulated in our previous study.41
However, only a small amount of high molecular weight PA8F was produced via the solid-state polymerization, suggesting that the efficiency of the enzymatic solid-state polymerization was quite low. As the cumulative weight fractions determined by SEC shown (see Fig. 4 and Table S2†), the enzymatic solid-state polymerization afforded ∼3–4% of additional PA8F with  of 42–509 (5500–67200 g mol−1) and ∼5% of extra PA8F with higher
of 42–509 (5500–67200 g mol−1) and ∼5% of extra PA8F with higher  of 509–1350 (67200–178400 g mol−1), when the polymerization time was increased from 24 to 72 h.
of 509–1350 (67200–178400 g mol−1), when the polymerization time was increased from 24 to 72 h.
We also found that a large proportion of short chain PA8F was produced via the enzymatic polymerization. As shown in Fig. 4 and Table S2,† after 72 h polymerization, ∼27% of the obtained crude PA8F possessed a low  of less than 16 (<2100 g mol−1); and the amount of PA8F with
of less than 16 (<2100 g mol−1); and the amount of PA8F with  of less than 42 (<5500 g mol−1) reached ∼52%. The short chain PA8F possesses a high solubility in the used precipitants (1,4-dioxane and methanol) and therefore, they were washed away during the purification steps. Thus, only less than ∼50% of the products were collected after purification, and this could be improved by choosing other proper precipitants.
of less than 42 (<5500 g mol−1) reached ∼52%. The short chain PA8F possesses a high solubility in the used precipitants (1,4-dioxane and methanol) and therefore, they were washed away during the purification steps. Thus, only less than ∼50% of the products were collected after purification, and this could be improved by choosing other proper precipitants.
In summary, the enzymatic polymerization kinetics study indicated that phase separation of FDCA-based oligoamides/polyamides occurred in the early stage of polymerization, and that the resulting PA8F has undergone a subsequent enzymatic solid-state polymerization. However, due to the phase separation and the low efficiency of the enzymatic solid-state polymerization, a large proportion of short chain PA8F was produced, which led to the low yields of the purified products.
N435-catalyzed polycondensation of DMFDCA and various aliphatic diamines via a one-stage method in toluene
A series of FDCA-based semi-aromatic polyamides including PA4F, PA6F, PA8F, PA10F and PA12F was successfully synthesized via a one-stage enzymatic polycondensation in toluene (see Scheme 1). The diamines used were (potentially) biobased 1,4-butanediamine (1,4-BDA), 1,6-hexanediamine (1,6-HDA), 1,8-octanediamine (1,8-ODA), 1,10-decanediamine (1,10-DDA), and 1,12-dodecanediamine (1,12-DODA). The number of methylene units in the diamine segments is 4, 6, 8, 10 and 12, respectively, which defines the chain length (x) of the tested aliphatic diamines.The chemical structures of the obtained FDCA-based semi-aromatic polyamides are confirmed by NMR and ATR-FTIR (see Fig. 6 and 7). The detailed NMR and IR assignments are described in the Experimental section and in our previous report.41
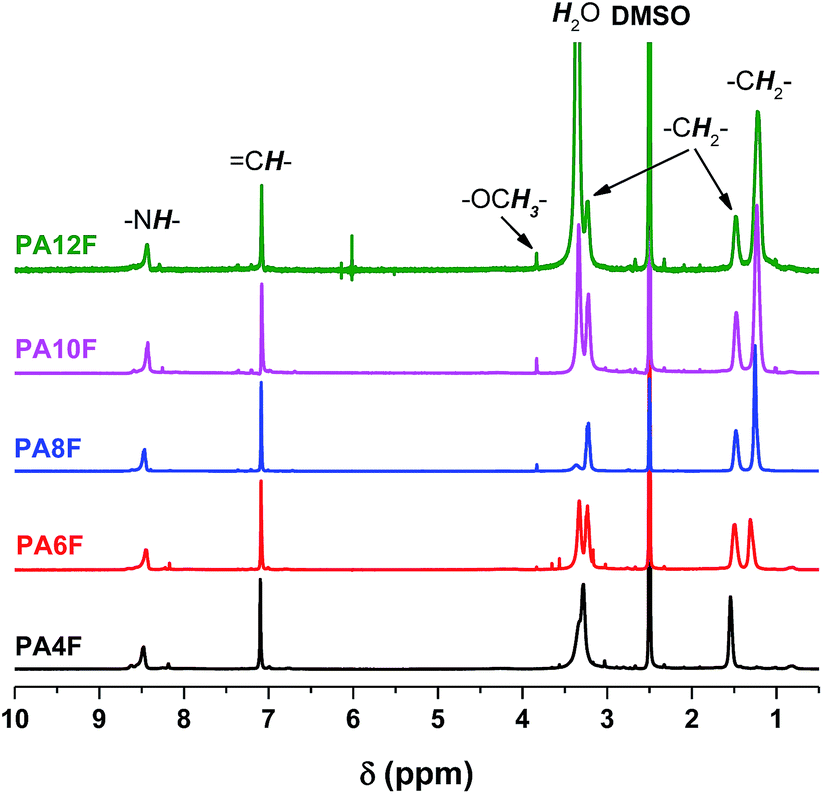 | ||
| Fig. 6 1H NMR spectra of FDCA-based semi-aromatic polyamides. | ||
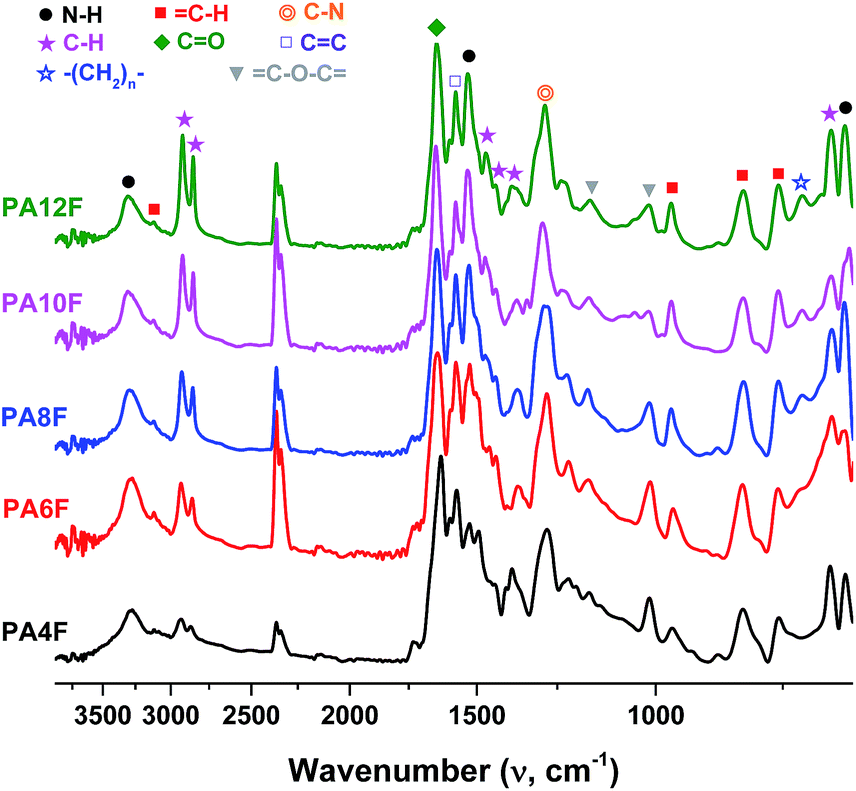 | ||
| Fig. 7 ATR-FTIR spectra of FDCA-based semi-aromatic polyamides. | ||
Effect of diamine chain length on enzymatic polymerization
The molecular weights of the obtained FDCA-based semi-aromatic polyamides were determined by NMR and SEC, as summarized in Tables 1 and S3 in the ESI.† The SEC retention curves are illustrated in Fig. S1 (see ESI†) and in our previous report.41
Fig. 8 depicts  and yield of the purified FDCA-based semi-aromatic polyamides against the chain length of the tested aliphatic diamines. The same trend with respect to the diamine chain length is observed: the
and yield of the purified FDCA-based semi-aromatic polyamides against the chain length of the tested aliphatic diamines. The same trend with respect to the diamine chain length is observed: the  and yield of the purified products increased significantly with increasing the diamine chain length from 4 to 8; however, by further increasing the diamine chain length from 8 to 12, the enzymatic polymerization resulted in a lower
and yield of the purified products increased significantly with increasing the diamine chain length from 4 to 8; however, by further increasing the diamine chain length from 8 to 12, the enzymatic polymerization resulted in a lower  and a reduction in yield of the purified products. This indicated that N435 shows the highest selectivity towards 1,8-ODA, which is in good agreement with the previous study on the enzymatic polymerization of aliphatic polyamides as reported by our laboratory.30
and a reduction in yield of the purified products. This indicated that N435 shows the highest selectivity towards 1,8-ODA, which is in good agreement with the previous study on the enzymatic polymerization of aliphatic polyamides as reported by our laboratory.30
 | ||
Fig. 8
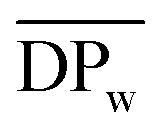 and isolation yield of the purified FDCA-based semi-aromatic polyamides from the enzymatic polymerization against the diamine chain length. and isolation yield of the purified FDCA-based semi-aromatic polyamides from the enzymatic polymerization against the diamine chain length. | ||
The enzymatic polymerization with 1,4-BDA having the shortest chain length amongst the tested aliphatic diamines resulted in PA4F with the lowest isolation yield. This could be mainly attributed to two reasons. First, 1,4-BDA is not favored by CALB due to its short chain length while second PA4F possesses the lowest solubility among the FDCA-based semi-aromatic polyamides.
Moreover, the isolation yield of the purified products from the enzymatic polymerizations was less than ∼50%. This can be explained by the production of a large proportion of low molecular weight oligoamides due to the occurrence of phase separation and the low efficiency of the enzyme-catalyzed solid-state polymerization as discussed before. Noteworthy is that the enzymatic polymerization can be significantly improved by using a two-stage method in diphenyl ether as reported in our previous study.41 We found that the two-stage enzymatic polymerization in diphenyl ether can afford high molecular weight PA8F with a higher isolation yield of ∼70%.
Enzymatic approach versus conventional synthesis techniques
Previously, FDCA-based semi-aromatic polyamides were produced via conventional synthesis techniques, including melt, solution and interfacial polymerization, as reported in literature.42–44 Due to the low solubility of the resulting polyamides and the extensive occurrence of side-reactions at elevated temperatures such as the decarboxylation of FDCA and N-methylation of (poly)amides,8 FDCA-based semi-aromatic polyamides with relatively low molecular weights were obtained, with and
and  ranged from 4000 to 10000 and 11100 to 18800 g mol−1, respectively (Table 1). However, in our study, even under a mild reaction temperature, the enzymatic polymerization generally resulted in a higher
ranged from 4000 to 10000 and 11100 to 18800 g mol−1, respectively (Table 1). However, in our study, even under a mild reaction temperature, the enzymatic polymerization generally resulted in a higher 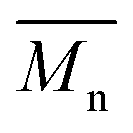 and
and 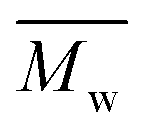 of around 9500–13400 and 15800–48300 g mol−1, respectively. As confirmed by MALDI-ToF MS analysis (see below), no side-products were formed during the enzymatic polymerization, which could have indicated the decarboxylation of DMFDCA and N-methylation of (poly)amides. This suggested that such side-reactions were greatly prevented by using N435 as the catalyst. To conclude, we have proven that enzymatic catalysis is a powerful approach towards the synthesis of sustainable FDCA-based semi-aromatic polyamides.
of around 9500–13400 and 15800–48300 g mol−1, respectively. As confirmed by MALDI-ToF MS analysis (see below), no side-products were formed during the enzymatic polymerization, which could have indicated the decarboxylation of DMFDCA and N-methylation of (poly)amides. This suggested that such side-reactions were greatly prevented by using N435 as the catalyst. To conclude, we have proven that enzymatic catalysis is a powerful approach towards the synthesis of sustainable FDCA-based semi-aromatic polyamides.
| Polymer | Synthesis approach |
|
|
T m (°C) | ΔHm (J g−1) | T g (°C) | T d (°C) |
|---|---|---|---|---|---|---|---|
a
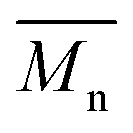 (number-average molecular weight) and (number-average molecular weight) and 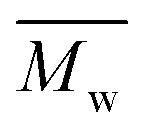 (weight-average molecular weight) were determined by SEC using DMF/LiBr as the eluent.
b (weight-average molecular weight) were determined by SEC using DMF/LiBr as the eluent.
b
 and and  were determined by SEC using DMSO/LiBr as the eluent.
c were determined by SEC using DMSO/LiBr as the eluent.
c
 was determined by end group titration.
d was determined by end group titration.
d
 and and  were determined by SEC using HFIP as the eluent.
e Not mentioned in the literature.
f Not detected at the tested time temperature scales.
g
T
m (melting temperature) and ΔHm (enthalpy of fusion) were determined by DSC (first heating scan), with a heating rate of 10 °C min−1; the Tg (glass transition temperature) was determined by DSC (second heating scan), with a heating rate of 10 °C min−1.
h The test method wass not mentioned in the literature.
i
T
m and Tg were determined by DTA (differential thermal analysis) at a heating rate of 20 °C min−1.
j
T
g was determined by DSC at a heating rate of 5 °C min−1.
k Decomposition temperature at 5% weight loss (Td-5%).
l Temperature at the maximum rate of decomposition (Td-max).
m Initial degradation temperature.
n Framework collapses temperature; Td = decomposition temperature. were determined by SEC using HFIP as the eluent.
e Not mentioned in the literature.
f Not detected at the tested time temperature scales.
g
T
m (melting temperature) and ΔHm (enthalpy of fusion) were determined by DSC (first heating scan), with a heating rate of 10 °C min−1; the Tg (glass transition temperature) was determined by DSC (second heating scan), with a heating rate of 10 °C min−1.
h The test method wass not mentioned in the literature.
i
T
m and Tg were determined by DTA (differential thermal analysis) at a heating rate of 20 °C min−1.
j
T
g was determined by DSC at a heating rate of 5 °C min−1.
k Decomposition temperature at 5% weight loss (Td-5%).
l Temperature at the maximum rate of decomposition (Td-max).
m Initial degradation temperature.
n Framework collapses temperature; Td = decomposition temperature.
|
|||||||
| PA4F | Enzymatic | 12300a |
15800 |
—f | —f | 119g | 257k; 443l |
| PA6F | Enzymatic | 13400a |
20600 |
162g | 6g | 119g | 322k; 460l |
| PA8F | Enzymatic | 13400b |
48300 |
160g | 10g | 118g | 378k; 464l |
| PA10F | Enzymatic | 13400a |
21200 |
135g | 47g | 98g | 366k; 463l |
| PA12F | Enzymatic | 9500a | 19900 |
125138g |
43g | 82g | 321k; 466l |
| PA4F43 | Interfacial | 6000–10000c |
—e | 250h | —e | —e | —e |
| PA6F44 | Melt | 5200d | 13800 |
—e | —e | 110j | 350–450h |
| PA6F43 | Interfacial/solution/melt | 6000–10000c |
—e | 155–175h | —e | —e | —e |
| PA8F44 | Melt | 4300d | 11100 |
—e | —e | 83j | 350–450h |
| PA8F43 | Melt | 6000–10000c |
—e | 125h | —e | —e | —e |
| PA10F44 | Melt | 5300d | 12700 |
—e | —e | 71j | 350–450h |
| PA10F42 | Interfacial | ≤8800 ( ≤ 30)h ≤ 30)h |
—e | 130h | —e | — | — |
| PA12F44 | Melt | 7000d | 18800 |
—e | —e | 68j | 350–450h |
| PA4T49 | Interfacial | —e | —e | 436i | —e | —e | —e |
| PA4T50 | —e | —e | —e | 430g | —e | —e | 350m |
| PA6T49 | Interfacial | —e | —e | 371i | —e | 125i | —e |
| PA6T50 | —e | —e | —e | 370g | —e | —e | 350m |
| PA8T49 | Interfacial | —e | —e | 335i | —e | 123i | —e |
| PA8T51 | —e | —e | —e | 338g | —e | —e | 402m |
| PA10T52 | Solid-state | —e | —e | 290g | —e | 113g | ∼426m; ∼482n |
| PA12T52 | Solid-state | —e | —e | 270g | —e | 96g | ∼426m; ∼482n |


It should be noted that the reaction time used for the enzymatic polymerization was longer than that applied for the conventional synthesis approaches. However, as indicated by the enzymatic kinetics study, the enzymatic polymerization time could be shortened from 72 to ∼24 h. In this case the enzymatic polymerization could result in FDCA-based semi-aromatic polyamides with similar  values. Therefore, the enzymatic polymerization is comparable to the conventional approaches with respect to the reaction time and the resulting molecular weights. Furthermore, the enzymatic polymerization is an energy saving process as the reaction temperature is mild; and no harmful residuals are remained in the resulting polyamides by using enzymes as catalysts. Besides, the immobilized enzyme catalysts can be recycled and reused for many cycles.55
values. Therefore, the enzymatic polymerization is comparable to the conventional approaches with respect to the reaction time and the resulting molecular weights. Furthermore, the enzymatic polymerization is an energy saving process as the reaction temperature is mild; and no harmful residuals are remained in the resulting polyamides by using enzymes as catalysts. Besides, the immobilized enzyme catalysts can be recycled and reused for many cycles.55
MALDI-ToF MS analysis
The end groups of the obtained FDCA-based semi-aromatic polyamides were determined by MALDI-ToF MS. Eight polyamide species with different abundance were identified (see Table 2, Fig. 9, and S2–S5 in the ESI†), which is in good agreement with our previous study.41 The identified polyamide species were terminated with ester/amine, ester/ester, amine/amine, acid/amine, ester/acid, acid/acid, and ester/amide, as well as, without end groups (cyclic polyamides).| Entry | Polymer species | End groups | Remaining mass (amu) |
|---|---|---|---|
| A |

|
Ester/amine | 32.03 |
| B |

|
Ester/ester | 184.15 |
|
x = 4:88.15 |
|||
|
x = 6:116.21 |
|||
| C |
|
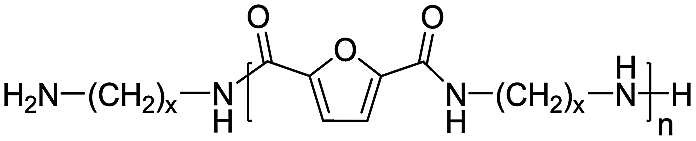
Amine/amine |
x = 8:114.26 |
|
x = 10:172.32 |
|||
|
x = 12:200.37 |
|||
| D |
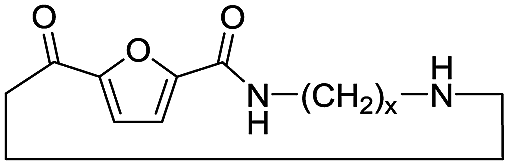
|
Cyclic | 0 |
| E |

|
Acid/amine | 18.02 |
| F |
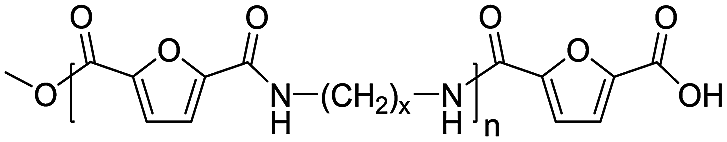
|
Ester/acid | 170.12 |
| G |
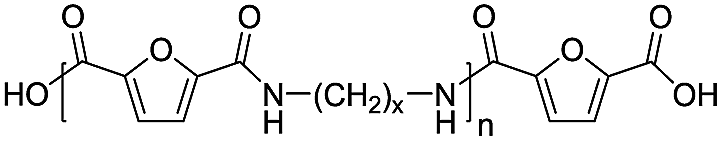
|
Acid/acid | 156.09 |
| H |

|
Ester/amide | 60.05 |
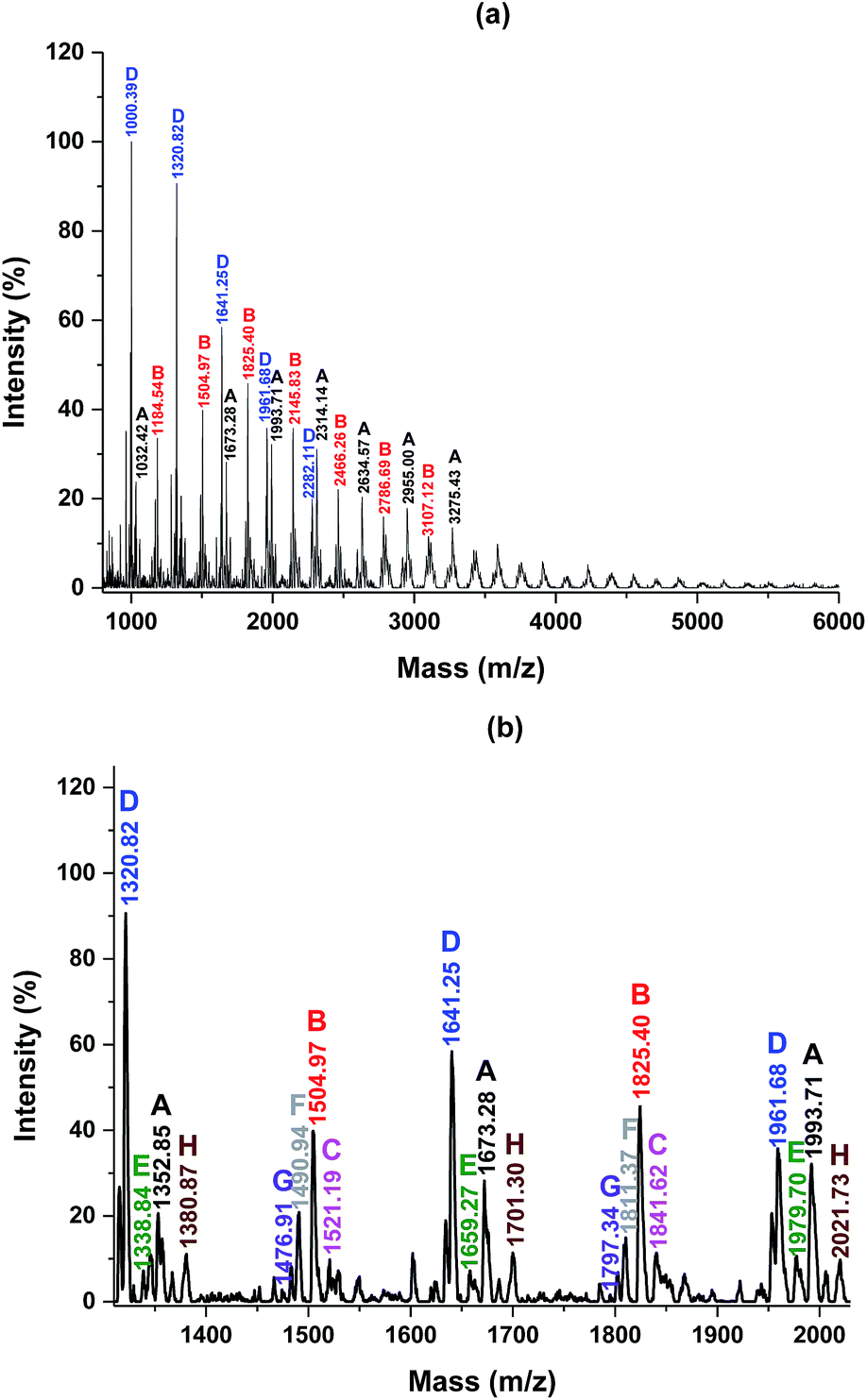 | ||
| Fig. 9 MALDI-ToF MS spectrum of the obtained PA12F with detailed peak interpretation. | ||
The acid end group was produced by a N435-catalyzed hydrolysis of esters during the polymerization;31,41 and the formation of the amide end group was due to the reaction between the amine groups and formic acid at the purification step.31,41,56
Thermal properties of the obtained FDCA-based semi-aromatic polyamides
The thermal stability of the obtained FDCA-based semi-aromatic polyamides was characterized by TGA. All tested polyamides showed a decomposition temperature at 5% weight loss (Td-5%) and a temperature at the maximum rate of decomposition (Td-max) at around 257–378 and 443–466 °C, respectively (see Table 1 and Fig. 10). We also found that the obtained PA4F is less thermally stable compared to the other tested polymers, which is probably due to its relatively low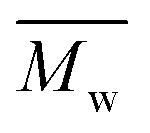 . Moreover, considering their high Td-max, all the tested FDCA-based semi-aromatic polyamides possess a very broad processing window.
. Moreover, considering their high Td-max, all the tested FDCA-based semi-aromatic polyamides possess a very broad processing window.
 | ||
| Fig. 10 TGA curves of the obtained FDCA-based semi-aromatic polyamides produced via a one-stage enzymatic polymerization in toluene at 90 °C. | ||
The thermal transitions of the obtained FDCA-based semi-aromatic polyamides were analyzed by DSC (see Table 1, Fig. 11, and S6–S9 in the ESI†). Broad melting peaks ranging from 125 to 162 °C were observed in the first DSC heating curves, except for PA4F which possesses a high Tm above the tested temperature (200 °C) due to its high chain rigidity. As reported by Heertjes and Kok,43 the Tm of PA4F with a  of around 6000–10000 g mol−1 is 250 °C. Moreover, the tested FDCA-based semi-aromatic polyamides possessed a lower Tm when the chain length of aliphatic diamine units increased (Fig. 12a). A similar trend was also observed for the FDCA-based semi-aromatic polyesters40 and TPA-based semi-aromatic polyamides.50,52 In this study, the decrease of Tm with increasing of the aliphatic diamine chain length can be explained by two facts. The increase of the aliphatic diamine chain length results in an increase in the chain flexibility and a decrease in the density of hydrogen bonds and π–π stackings.
of around 6000–10000 g mol−1 is 250 °C. Moreover, the tested FDCA-based semi-aromatic polyamides possessed a lower Tm when the chain length of aliphatic diamine units increased (Fig. 12a). A similar trend was also observed for the FDCA-based semi-aromatic polyesters40 and TPA-based semi-aromatic polyamides.50,52 In this study, the decrease of Tm with increasing of the aliphatic diamine chain length can be explained by two facts. The increase of the aliphatic diamine chain length results in an increase in the chain flexibility and a decrease in the density of hydrogen bonds and π–π stackings.
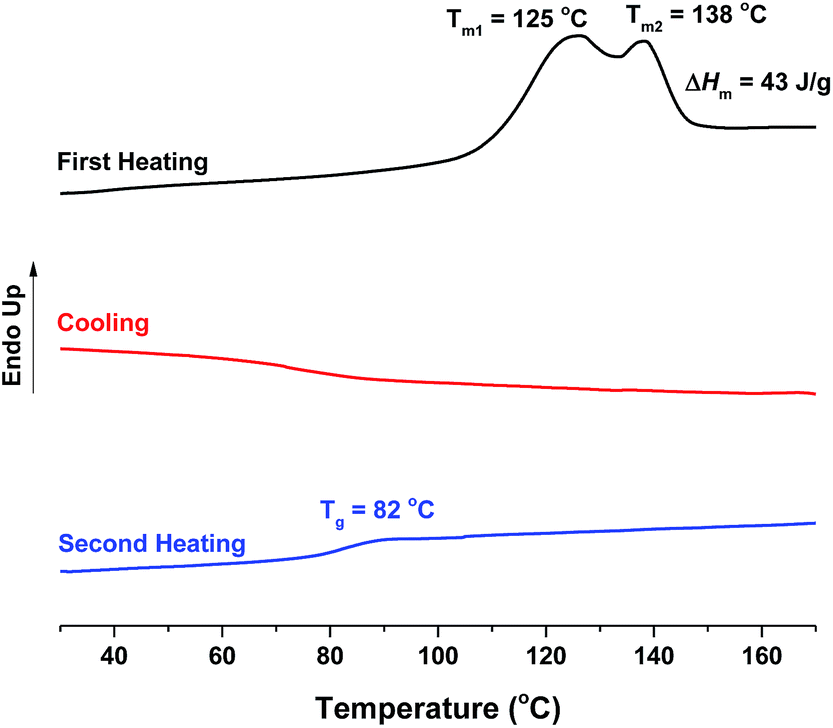 | ||
Fig. 11 DSC curves of the obtained PA12F. PA12F ( = 9500 g mol−1, = 9500 g mol−1,  = 19900 g mol−1) was produced via a one-stage enzymatic polymerization of DMFDCA and 1,12-DODA in toluene at 90 °C. = 19900 g mol−1) was produced via a one-stage enzymatic polymerization of DMFDCA and 1,12-DODA in toluene at 90 °C. | ||
 | ||
| Fig. 12 (a) Tm, and (b) Tg of the FDCA-based semi-aromatic polyamides (PAXF, X represents the chain length of aliphatic diamine units) and TPA-based semi-aromatic polyamides (PAXT). The FDCA-based semi-aromatic polyamides were produced via enzymatic polymerizations in this study or by conventional synthesis techniques as reported in the literature,42–44 while the TPA-based semi-aromatic polyamides were synthesized via conventional approaches as reported in the literature.49–52 If the polyamides possess several melting peaks or showed different Tm in the literature, the highest Tm is presented. | ||
The enthalpy of fusion (ΔHm) of the tested FDCA-based semi-aromatic polyamides increased significantly from 6 to ∼40 J g−1 when the chain length of aliphatic diamine units was increased from 6 to 10 and 12 (see Table 1). This may suggest that the FDCA-based semi-aromatic polyamides with longer aliphatic diamine units possess a higher crystallization ability because of the higher chain flexibility.
No crystallization was observed in the DSC cooling curves of all tested polyamides, and no melting peak appeared in the second heating curves (see Fig. 11, and S6–S9 in the ESI†). This indicated that all synthesized FDCA-based semi-aromatic polyamides cannot crystallize in bulk at the tested conditions, which can be explained by their slow crystallization rate.41
The Tg of the obtained FDCA-based semi-aromatic polyamides decreased gradually from 119 to 82 °C when the chain length of aliphatic diamine units was increased from 4 to 12 (Fig. 12b), which can be explained by the same line of reasoning as discussed above for Tm. Again this agreed well with the previous studies reported.40,52,57 It was found that the FDCA-based semi-aromatic polyesters and TPA-based semi-aromatic polyamides showed a continuous reduction in Tg with an increase of the chain length of aliphatic diol/diamine units.
Comparison of the thermal properties: enzymatic FDCA-based semi-aromatic polyamides versus FDCA-based and TPA-based counterparts produced via conventional synthesis techniques
The FDCA-based semi-aromatic polyamides produced from the enzymatic polymerizations possess similar thermal stability compared to the FDCA-based and TPA-based counterparts obtained via conventional approaches (see Table 1).Moreover, Tm and Tg of the enzymatic FDCA-based semi-aromatic polyamides are generally higher than those of the counterparts synthesized via the conventional synthesis techniques (Fig. 12). This could be rationalized by the higher molecular weights of the enzymatic FDCA-based polyamides.
Furthermore, Tg of the enzymatic FDCA-based semi-aromatic polyamides is a bit lower than that of the TPA-based counterparts. This could be attributed to the lower molecular weights of the FDCA-based semi-aromatic polyamides, and the lower density of intermolecular hydrogen bonds. The latter is due to the formation of additional interactions between amide groups and oxygen heteroatoms of the furan ring as reported in literature.58 Given the fact that the difference in Tg is quite small (6–15 °C), we can draw the conclusion that the FDCA-based and TPA-based semi-aromatic polyamides possess a comparable Tg.
However, the TPA-based semi-aromatic polyamides possess a much higher Tm, which is more than 130 °C higher than those of the FDCA-based counterparts. This is attributed to the formation of defected polymer crystals in the FDCA-based semi-aromatic polyamides and their lower degree of crystallinity, which is caused by the perturbed hydrogen bondings due to the additional interactions between amide groups and oxygen heteroatoms of furan rings.58
Conclusions
A series of FDCA-based semi-aromatic polyamides is successfully synthesized via N435-catalyzed polycondensation of (potentially) biobased DMFDCA and aliphatic diamine differing in chain length (C4–C12), using a one-stage method at 90 °C in toluene.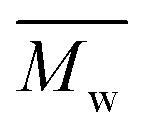 of the resulting products ranges from 15800 to 48300 g mol−1, which is higher than those produced via conventional synthesis techniques. We found that N435 shows the highest selectivity towards 1,8-ODA (C8) among the tested aliphatic diamines. Moreover, MALDI-ToF MS analysis suggested that no byproducts were produced, which could have indicated the occurrence of undesirable side-reactions during the enzymatic polymerization. Therefore, enzymatic catalysis is proven to be a robust pathway towards the synthesis of sustainable FDCA-based semi-aromatic polyamides. These FDCA-based semi-aromatic polyamides are promising biobased substitutes to petrol-based counterparts, which have a great commercial interest as thermal engineering plastics and high performance materials.
of the resulting products ranges from 15800 to 48300 g mol−1, which is higher than those produced via conventional synthesis techniques. We found that N435 shows the highest selectivity towards 1,8-ODA (C8) among the tested aliphatic diamines. Moreover, MALDI-ToF MS analysis suggested that no byproducts were produced, which could have indicated the occurrence of undesirable side-reactions during the enzymatic polymerization. Therefore, enzymatic catalysis is proven to be a robust pathway towards the synthesis of sustainable FDCA-based semi-aromatic polyamides. These FDCA-based semi-aromatic polyamides are promising biobased substitutes to petrol-based counterparts, which have a great commercial interest as thermal engineering plastics and high performance materials.
The enzymatic polymerization kinetics study suggested that  increases significantly with increase of polymerization time. We also found that phase separation occurs in the early stage of polymerization, and the enzymatic solid-state polymerization takes place. However, a large proportion of the resulting polyamides possess low molecular weights. As a result, only less than ∼50% of the products were obtained after purification.
increases significantly with increase of polymerization time. We also found that phase separation occurs in the early stage of polymerization, and the enzymatic solid-state polymerization takes place. However, a large proportion of the resulting polyamides possess low molecular weights. As a result, only less than ∼50% of the products were obtained after purification.
All obtained FDCA-based semi-aromatic polyamides possess a Td-5% and Td-max at around 257–378 and 443–466 °C, respectively. In addition, these enzymatic polyamides display a Tm of around 125–162 °C (except PA4F), and a Tg of around 82–119 °C. Moreover, the tested enzymatic FDCA-based semi-aromatic polyamides having longer diamine units generally possess a lower Tm and Tg, but a higher crystallization ability.
The enzymatic FDCA-based semi-aromatic polyamides show a slightly higher Tm and Tg, and similar thermal stability compared to those produced via conventional synthesis techniques. Furthermore, these enzymatic FDCA-based semi-aromatic polyamides possess a comparable Tg and thermal stability, but a much lower Tm compared to the TPA-based counterparts.
Considering the fact that toluene is not an eco-friendly solvent, our future research will focus on the use of non-solvent and green solvents such as ionic liquids and supercritical CO2 for the enzymatic polymerization of biobased polyamides. We also hope that the isolation yield and molecular weights can be improved by using such green solvents.
Acknowledgements
This research forms part of the research programme of the Dutch Polymer Institute (DPI), project #727c polymers go even greener. The authors thank Masyitha Ambarwati for the photograph in the graphic table of contents. Dina Maniar thanks the financial support from the Indonesia Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan/LPDP).Notes and references
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- I. Delidovich, P. J. Hausoul, L. Deng, R. Pfutzenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef CAS PubMed.
- A. Scott, Chem. Eng. News, 2016, 94, 15 Search PubMed.
- E. D. Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, vol. 1105, ch. 1, pp. 1–13 Search PubMed.
- P. F. H. Harmsen, M. M. Hackmann and H. L. Bos, Biofuels, Bioprod. Biorefin., 2014, 8, 306–324 CrossRef CAS.
- A. Gandini, T. M. Lacerda, A. J. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- M. Trigo-López, P. Estévez, N. San-José, A. Gómez-Valdemoro, F. C. García, F. Serna, J. L. Pena and J. M. García, Recent Pat. Mater. Sci., 2009, 2, 190–208 CrossRef.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef.
- K. Marchildon, Macromol. React. Eng., 2011, 5, 22–54 CrossRef CAS.
- J. I. Iribarren, C. Alemán and J. Puiggalí, in Handbook of Engineering and Specialty Thermoplastics, John Wiley & Sons, Inc., 2011, ch. 3, pp. 43–77, DOI:10.1002/9781118229064.ch3.
- S. Kobayashi, S. Shoda and H. Uyama, in Polymer Synthesis/Polymer Engineering, Springer-Verlag, Berlin Heidelberg, 1995, vol. 121, pp. 1–30 Search PubMed.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- N. Miletić, K. Loos and R. A. Gross, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, ch. 4, pp. 83–129, DOI:10.1002/9783527632534.ch4.
- J. X. Zhang, H. Shi, D. Wu, Z. Xing, A. J. Zhang, Y. Yang and Q. S. Li, Process Biochem., 2014, 49, 797–806 CrossRef CAS.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- S. Kobayashi, Polym. Adv. Technol., 2015, 26, 677–686 CrossRef CAS.
- K. Loos, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 1–29 Search PubMed.
- A. Singh and D. L. Kaplan, in Enzyme-Catalyzed Synthesis of Polymers, ed. S. Kobayashi, H. Ritter and D. Kaplan, Springer-Verlag, Berlin Heidelberg, 2006, vol. 194, pp. 211–224 Search PubMed.
- F. Hollmann, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, ch. 6, pp. 143–163, DOI:10.1002/9783527632534.ch6.
- J. van der Vlist and K. Loos, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, ch. 9, pp. 211–246, DOI:10.1002/9783527632534.ch9.
- E. Stavila and K. Loos, J. Renewable Mater., 2015, 3, 268–280 CrossRef.
- G. G. Linares and A. Baldessari, Curr. Org. Chem., 2013, 17, 719–743 CrossRef CAS.
- H. N. Cheng, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, ch. 5, pp. 131–141, DOI:10.1002/9783527632534.ch5.
- E. Stavila, R. Z. Arsyi, D. M. Petrovic and K. Loos, Eur. Polym. J., 2013, 49, 834–842 CrossRef CAS.
- E. Stavila, G. O. R. Alberda van Ekenstein and K. Loos, Biomacromolecules, 2013, 14, 1600–1606 CrossRef CAS PubMed.
- K. Brandstadt, T. Lane and R. Gross, US Pat. US20070021578 A1, 2006.
- G. Qu-Ming, W. W. Maslanka and H. N. Cheng, in Polymer Biocatalysis and Biomaterials II, American Chemical Society, Washington, DC, 2008, vol. 999, ch. 21, pp. 309–319 Search PubMed.
- L. Ragupathy, U. Ziener, R. Dyllick-Brenzinger, B. von Vacano and K. Landfester, J. Mol. Catal. B: Enzym., 2012, 76, 94–105 CrossRef CAS.
- F. Poulhès, D. Mouysset, G. Gil, M. P. Bertrand and S. Gastaldi, Polymer, 2013, 54, 3467–3471 CrossRef.
- I. Baum, B. Elsässer, L. W. Schwab, K. Loos and G. Fels, ACS Catal., 2011, 1, 323–336 CrossRef CAS.
- Y. Poojari and S. J. Clarson, Macromolecules, 2010, 43, 4616–4622 CrossRef CAS.
- E. Stavila, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Biomacromolecules, 2014, 15, 234–241 CrossRef CAS PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrovic and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5198–5211 RSC.
- Y. Jiang, D. Maniar, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Biomacromolecules, 2015, 16, 3674–3685 CrossRef CAS PubMed.
- H. Hopff and A. Krieger, Makromol. Chem., 1961, 47, 93–113 CrossRef CAS.
- P. Heertjes and G. Kok, Delft Prog. Rep., Ser. A, 1974, 1, 59–63 CAS.
- O. Grosshardt, U. Fehrenbacher, K. Kowollik, B. Tubke, N. Dingenouts and M. Wilhelm, Chem. Ing. Tech., 2009, 81, 1829–1835 CrossRef.
- A. Duursma, R. Aberson, D. D. Smith, J. Flores, M. A. Dam and G. J. M. Gruter, International Patent WO 2015060718 A1, 2015.
- D. D. Smith, J. Flores, R. Aberson, M. A. Dam, A. Duursma and G. J. M. Gruter, International Patent WO 2015059047 A1, 2015.
- V. N. Tsarev, Y. Morioka, J. Caner, Q. Wang, R. Ushimaru, A. Kudo, H. Naka and S. Saito, Org. Lett., 2015, 17, 2530–2533 CrossRef CAS PubMed.
- H. Hopff and A. Krieger, Helv. Chim. Acta, 1961, 44, 1058–1063 CrossRef CAS.
- P. W. Morgan and S. L. Kwolek, Macromolecules, 1975, 8, 104–111 CrossRef CAS.
- G. Zhang, Y. X. Zhou, Y. Li, X. J. Wang, S. R. Long and J. Yang, RSC Adv., 2015, 5, 49958–49967 RSC.
- G. Zhang, H. W. Yang, S. X. Zhang, Y. Zhang, X. J. Wang and J. Yang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2012, 49, 414–423 CrossRef CAS.
- W. Z. Wang and Y. H. Zhang, eXPRESS Polym. Lett., 2009, 3, 470–476 CrossRef CAS.
- Y. Jiang, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Macromol. Chem. Phys., 2014, 215, 2185–2197 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Biomolecules, 2013, 3, 461–480 CrossRef PubMed.
- L. W. Ren, Y. S. Wang, J. Ge, D. N. Lu and Z. Liu, Macromol. Chem. Phys., 2015, 216, 636–640 CrossRef CAS.
- A. R. Katritzky, R. L. Parris, E. S. Ignatchenko, S. M. Allin and M. Siskin, J. Prakt. Chem./Chem.-Ztg., 1997, 339, 59–65 CrossRef CAS.
- J. Wu, P. Eduard, S. Thiyagarajan, L. Jasinska-Walc, A. Rozanski, C. F. Guerra, B. A. J. Noordover, J. van Haveren, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 5069–5080 CrossRef CAS.
- C. H. R. M. Wilsens, Y. S. Deshmukh, B. A. J. Noordover and S. Rastogi, Macromolecules, 2014, 47, 6196–6206 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular weights of the crude PA8F from the enzymatic kinetics study, cumulative weight fractions of the crude PA8F determined by SEC, molecular weights and isolation yields of the purified FDCA-based semi-aromatic polyamides, SEC elution curves of the obtained FDCA-based semi-aromatic polyamides, MALDI-ToF MS spectra and DSC curves of the obtained FDCA-based semi-aromatic polyamides. See DOI: 10.1039/c6ra14585j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |