
Synthesis of cinchona alkaloid sulfonamide polymers as sustainable catalysts for the enantioselective desymmetrization of cyclic anhydrides†
Abstract
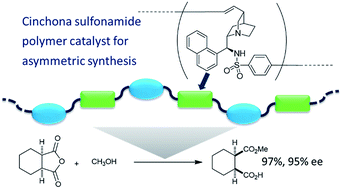
The Mizoroki–Heck polymerization of cinchona-based sulfonamide dimers and aromatic diiodides was investigated in the presence of a palladium catalyst, to obtain chiral polymers in high yields. An iodobenzenesulfonamide derivative of a cinchona alkaloid was also polymerized via self-polycondensation under the same reaction conditions. The catalytic activities of these chiral polymers were examined by using them as catalysts in the enantioselective desymmetrization of cyclic anhydrides.
 Please wait while we load your content...
Please wait while we load your content...

