
Identification of novel pyrazole–rhodanine hybrid scaffolds as potent inhibitors of aldose reductase: design, synthesis, biological evaluation and molecular docking analysis†

Abstract
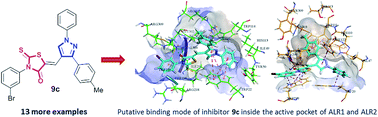
In an effort to develop a new class of potent aldose reductase inhibitors, a series of 1,3-diarylpyrazole assimilated 3-substituted 4-oxo-2-thioxo-1,3-thiazolidines (9a–n) was designed, and synthesized in good to excellent yields by a pharmacophore integration approach. The structures of the newly synthesized pyrazole–rhodanine derivatives were established by readily available spectroscopic methods (FTIR, 1H and 13C NMR) and mass spectrometry. The hybrid compounds were evaluated as aldehyde and aldose reductase inhibitors. The biological screening results identified several compounds as remarkable inhibitors of ALR1 and ALR2. Among them, compounds 9c and 9k showed excellent activity (and complete selectivity) towards the aldose reductase enzyme with IC50 values of 1.22 ± 0.67, and 2.34 ± 0.78 μM, respectively, as compared to the standard drug (sorbinil; IC50 = 3.10 ± 0.20 μM). The molecular docking analysis of the most potent inhibitor 9c was performed in order to identify the putative binding modes inside the active pocket of the enzymes. These newly discovered aldose reductase inhibitors are believed to represent valuable lead structures to further streamline the generation of candidate compounds to target a number of pathological conditions, most strikingly long-term diabetic complications.
 Please wait while we load your content...
Please wait while we load your content...

