
α-Iminonitrile: a new cyanating agent for the palladium catalyzed C–H cyanation of arenes†
Abstract
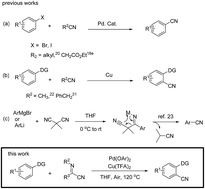
An efficient palladium-catalyzed C–H cyanation reaction of arenes using α-iminonitrile as a new cyanating reagent has been developed. With high regioselectivity and broad substrate scope, this reaction offers monocyanated products in moderate to excellent yields.
- This article is part of the themed collection: Editors’ collection: Catalytic Organic Transformations
 Please wait while we load your content...
Please wait while we load your content...

