
High catalytic activity for CO oxidation on single Fe atom stabilized in graphene vacancies†
Yanan Tang*,
Jincheng Zhou,
Zigang Shen,
Weiguang Chen,
Chenggang Li and
Xianqi Dai*
Quantum Materials Research Center, College of Physics and Electronic Engineering, Zhengzhou Normal University, Zhengzhou 450044, China. E-mail: yntang2010@163.com; xqdaizs@163.com; Fax: +86 371 65502273; Fax: +86 371 65501661; Tel: +86 371 65502273 Tel: +86 371 65501661
First published on 2nd September 2016
Abstract
Inspired by the recently discovered dynamics of single Fe atoms in graphene vacancies, we systemically examined the stable configurations, electronic structures, and catalytic activities of Fe-atom-embedded graphene substrates (including monovacancy graphene (MG) and divacancy graphene (DG)) by using first-principles calculations. We found that the doped Fe on the MG sheet (Fe/MG) is more stable than that on the DG sheet (Fe/DG). Doping with Fe atoms provides more transferred electrons to fill the vacancy defects of graphene and allows it to exhibit a more positive charge, which effectively regulates O2 and CO adsorption. Also, the degree of interactions between the reactants and substrates are connected to the reaction pathways and energy barriers. For the Fe/MG sheet, the low coadsorption energy of gas molecules can promote the catalytic reaction through the Langmuir–Hinshelwood (LH) mechanism. In comparison, the initial step for CO oxidation on the Fe/DG sheet is through the Eley–Rideal (ER) mechanism, which is an energetically more favorable process. Moreover, the more stable Fe/MG sheet is a much more efficient catalyst for CO oxidation at low temperature, because the sequential reaction processes (LH and ER) have low enough energy barriers. These results provide valuable guidance on selecting the metal dopant in graphene materials to design effective atomic-scale catalysts.
1. Introduction
In recent decades, the low temperature catalytic CO oxidation has attracted great interest due to its fundamental and important applications in the area of environmental protection and in avoiding electronic poisoning in fuel cells.1–3 Generally, CO oxidation has been considered as a vital prototype reaction for exploring the reactivity of metals in heterogeneous catalysis.4–6 Many strategies have been chosen by using supported noble metals or metal oxides7–9 as catalysts, such as Pt,2,10,11 Rh,12 Au,4,13–15 and Pd,3 which exhibit high activity for CO oxidation. Furthermore, some reports state that small metal nanoparticles or clusters (e.g., of Au,15,16 Pt,17 Pd,18 and Ag19) can exhibit exceptional catalytic activity for CO oxidation. However, noble metal catalysts are very costly and usually require high reaction temperatures; also the catalytic activities of metal nanoparticles can be strongly size- and shape-dependent, which limits their general use on a large scale. As a result, catalysts with higher activity and lower cost are urgently required in many practical applications.To solve these scientific issues, the catalytic activity of noble metals can be dramatically improved by reducing the size of the catalysts to nanoparticles20–23 and even to single atoms.24 Single-atom catalysts supported on various substrates have attracted attention due to their intrinsic high activity, stability, and low cost.25–28 Recently, Qiao et al. found that single-atom Pt loading on FeOx exhibited excellent catalytic activity toward CO oxidation.29 These results illustrated that the kind of single-metal atom and the choice of support substrates can affect the reactivity and stability of catalysts, especially two dimensional (2D) substrates, and consequently 2D materials have been the focus of much research due to their excellent physical and chemical properties.30 Compared to carbon-based nanomaterials, including carbon black and carbon nanotubes (CNTs), graphene is a 2D atomic crystal structure consisting of a single layer of sp2-hybridized carbon atoms31 and is now recognized as a promising substrate to support metal atoms due to its outstanding reaction mechanisms,32 and electronic33 and thermal34 properties for developing new catalysts.35 Moreover, graphene as a support has a large surface-to-volume ratio, which is a benefit for heterogeneous catalysts.36 Recently, small metal clusters supported on graphene substrates exhibited unusually high catalytic activity as electrodes in fuel cells.37–43 However, the supported nanoclusters usually incorporate several structural isomers and the easy occurrence of structural interconversion between the isomers can affect their stability and catalytic efficiency.44 Uniformly dispersed single-metal atoms on a graphene substrate are considered to be highly promising for application in heterogeneous catalysis.
Chemical doping has been confirmed to be an effective approach to tailor the properties of graphene.45–47 Earlier investigations showed that the substitutional dopants in graphene can control the size and degree of catalyst dispersion,48 as well as the stability of the catalysts to exhibit a high reactivity performance.49–52 Compared to nonmetal dopants in graphene substrates, the much stronger binding between carbon vacancies and metal atoms can effectively improve the structural stability,53–55 electronic structure, and magnetic properties.56–59 However, experimental studies in the literature have demonstrated that metallic impurities can originate from the growth process of graphite on metal substrates.60 Moreover, carbon vacancies can be deliberately introduced in graphene sheets by high energy atom/ion bombardment61 and then the carbon vacancies can be filled with the desired metal atom.62,63 These efficient atomic doping techniques provide a way to analyze the interaction between the introduced atoms and graphene sheets. Theoretically, single-atom Au–,64 Fe–,65 Cu–,66 Pt–,67 Pd–,68 and Al–embedded graphene69 systems have been predicted to be highly active catalysts for CO oxidation. These observations confirmed that the metallic impurities in graphene sheets could be a better candidate for improving the stability and the lifetime of supported catalysts, and that a single-atom catalyst with a low cost and high activity would have great potential application in catalysis.
In general, the degree of interaction between the active sites of catalysts and adsorption species is proportional to the binding energy and the reaction energy barriers.70 Among the noble metal-atoms–embedded graphene, Au–embedded graphene shows the best performance for CO oxidation.64 However, the adsorption energy of CO is larger than that of the O2 molecule, indicating that CO prefers to adsorb at the Au atom, which could prevent the continuous oxidation reaction. With reducing the consumption of noble metal atoms, it is necessary to exploit efficient non-noble metal (NNM) atoms supported on a graphene complex for CO oxidation. Previous studies have shown that some NNM-atoms-embedded graphene substrates exhibit high catalytic activity for CO oxidation,48,65,71 but there is a lack of comparative analysis of the activation barriers of different reaction mechanisms, such as for the Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) reactions. Compared to the noble metal atoms (e.g., Pt or Pd),68,72,73 we chose Fe as the dispersed single atom, because Fe is inexpensive, environmentally benign, readily available, and is richly abundant in the earth, and so it meets the requirements for a cheap, green, and efficient catalyst. Like in any other real material, structural defects do exist in graphene,74–76 and these can be useful for achieving new functionalities.77–80 Recently, Robertson et al. observed that mobile Fe atoms resident on the mono- and divacancies in graphene surfaces are stable in comparison to that on the graphene edge.81 Here, it is significant to evaluate the catalytic activity of single Fe atoms on defective graphene sheets (including the monovacancy and divacancy).
Inspired by earlier investigations, we selected monovacancy and divacancy graphene (MG and DG) as the supporting material and single Fe atoms as the catalysts to study the CO oxidation reaction. Based on our calculations, the MG and DG substrates act as support materials for the uniformly dispersed single-atom Fe catalyst (Fe–graphene includes Fe/MG and Fe/DG). In this study, the stable configurations, electronic structures, and magnetic properties of Fe/MG (or Fe/DG) toward reactive gases (CO and O2) were investigated by first-principles based on density functional theory (DFT). It was found that the CO oxidation reactions on the Fe/MG substrate have lower energy barriers (first through the LH reaction, followed by the ER reaction) as compared with those on the Fe/DG surface. In addition, we further investigated the ER reaction without an intermediate state (i.e., CO + O2 = CO2 + Oads) and then with the dissociation of O2 molecules as the initial step and the generated O atoms reacting with two CO molecules. To the best of our knowledge, few reports provide much systemic information about the catalytic activities behind CO oxidation on metal-embedded graphene systems (including MG and DG), and hence this report provides a valuable reference for designing graphene-based catalysis systems.
2. Computational model and methods
Spin-polarized DFT calculations were carried out by the Vienna ab initio simulation package (VASP)82,83 with the projector augmented wave (PAW) pseudo-potentials.84 The exchange-correlation functions were described with the generalized gradient approximation (GGA) in the form of the Perdew, Burke, and Ernzernhof (PBE) functional.85 The kinetic energy cutoff for the plane-wave basis set was chosen as 450 eV. The C 2s2p, Fe 3d4s, and O 2s2p states were treated as valence electrons. A modified hexagonal graphene ribbon with a 5 × 7 supercell was adopted, and the vacuum layer was set to 15 Å to avoid the interaction among mirror images. The calculated lattice constant for the graphene sheet was 2.47 Å, which nearly approximates to the experimental value of 2.46 Å.86 In order to improve the convergence of the states near the Fermi level, the Brillouin zone (BZ) integration was sampled using a 3 × 3 × 1 Γ-centered Monkhorst–Pack (MP) grid, while a Γ-centered MP grid of 15 × 15 × 1 was used for the final density of states (DOS) calculations.Bader charge analysis87 was used to evaluate the atomic charges and electron transfer in the reactions. Adsorption energies and site preferences for each type of gas molecules were tested on the Fe–graphene surfaces. The climbing image nudged elastic band method (CI-NEB)88–90 was employed to investigate the saddle points and minimum energy path (MEP) for the diffusion and dissociation of adatoms or for the reaction gases on the graphene substrates. The geometric optimization and the search for the transition states (TS) were tested by means of frequency calculations, while those with one imaginary frequency corresponded to the metastable states, which could be viewed as the MS. A number of intermediate images were constructed along the reaction pathways between the initial state (IS) or TS and the final state (FS), and the spring force between adjacent images was set as 5.0 eV Å−1. The images were optimized until the combined force on each atom was less than 0.02 eV Å−1. The energy barrier (Ebar) of each chemical reaction was calculated by the energy difference between the IS and TS in each chemical reaction.
The adsorption energy (Eads) was calculated using the formula Eads = EA + EB − EAB, where EA, EB, and EAB are the total energies of the optimized adsorbates in the gas molecules (A: O2, O, CO, and CO2), the clean Fe-embedded graphene substrates (B: Fe/MG and Fe/DG), and the adsorbate-substrate systems, respectively. With this definition, a higher Eads value means a stronger adsorption.
3. Results and discussion
3.1. Geometry stabilities and electronic properties
First, the stable geometries of Fe anchored on MG and DG substrates were investigated, and the corresponding adsorption energies, bond lengths, and transferred electrons are shown in Table 1. As shown in Fig. 1(a), single-atom Fe is embedded into the MG sheet, where one C atom is substituted by one Fe atom. The calculated Eads of the Fe dopant in graphene is 7.28 eV, and the Fe atom is moved out of the plane to obtain more space (1.20 Å) due to its atomic radius (1.30 Å) being larger than that of the carbon atom. The bond distance between the Fe atom and the neighboring carbon atoms is 1.76 Å, which is in agreement with the reported results.53,65 Based on the Bader charge analysis,87 the transferring electrons (1.07 e) move from the Fe atom to the MG sheet and form strong covalent bonds between the Fe atom and C atoms. As shown in Fig. 1(b), the stable configuration of Fe/DG was investigated after geometry optimization. Compared with the case of the Fe/MG system, the doped Fe atom provides more transferred electrons (1.33 e) to the DG system, the corresponding bond length of Fe–C is longer at 1.90 Å, and the Fe atom nearly retains the planar from of the pristine graphene. It was found that the doped Fe atom in the DG sheet exhibits less stability (6.47 eV) than that on the MG sheet (7.28 eV), yet they are much more stable than the same atom on pristine graphene (pri-graphene, 1.19 eV). In addition, the calculated small diffusion barrier (Ebar, 0.52 eV) indicates that the Fe adatom can easily diffuse on to pri-graphene. For the MG and DG sheets, the Fe adatom on graphene diffuses from the neighboring H site to its neighboring vacancy site with a small difference in Ebar (0.40 eV vs. 0.50 eV), while the inverse pathway needs to overcome a larger energy barrier (6.23 eV vs. 5.51 eV). These results indicate that the Fe adatoms can diffuse easily on the pri-graphene surface and prefer to be trapped at the vacancy site and appear as Fe dopants, since a surface reaction at ambient temperature may occur only when the energy barrier is smaller than the critical barrier of 0.91 eV.69 This observation confirms that Fe–graphene systems would be stable enough to be utilized in catalytic reactions. Therefore, it can be seen that the Fe dopants can uniformly disperse on graphene and are quite stable without a clustering formation.| Systems | Eads (eV) | d1 (Å) | Δq (e) | d2 (Å) | |
|---|---|---|---|---|---|
| Fe/MG | Fe | 7.28 | 1.76 | −1.07 | 1.20 |
| O2 | 1.88 | 1.40 | +0.81 | 1.86 | |
| CO | 1.13 | 1.15 | +0.31 | 1.88 | |
| Fe/DG | Fe | 6.47 | 1.90 | −1.33 | 0.00 |
| O2 | 1.80 | 1.34 | +0.53 | 1.96 | |
| CO | 1.77 | 1.16 | +0.30 | 1.78 | |
 | ||
| Fig. 1 Top and side views of the geometric structure for (a) Fe/MG and (b) Fe/DG sheets. Black and green balls represent the C and Fe atoms, respectively. (c) Spin-resolved total DOS (TDOS), partial DOS (PDOS), and local PDOS (LDOS) for Fe/MG (thick lines) and Fe/DG sheet (thin lines) systems. The vertical dotted line denotes the Fermi level. | ||
To gain more insight into the origin of the high stability of the Fe/MG and Fe/DG systems, we investigated the DOS plots, as shown in Fig. 1(c). We found that the electron states of the MG and DG substrates are strongly altered when an Fe atom is doped in these systems. The broadened partial DOS (PDOS) of the Fe 3d states overlap with the total DOS (TDOS) of the systems and the local PDOS (LDOS) of the C atoms around the Fermi level (EF), suggesting a strong interaction between the Fe atom and adjacent C atoms. Besides, the spin-up and spin-down channels of the Fe/MG (or Fe/DG) become asymmetric, indicating that these systems exhibit a magnetic character. As shown in Fig. 2, we also investigated the spin charge redistribution between Fe atoms and the MG (or DG) sheet. As shown in Fig. 2(a), more electrons dominantly accumulate in the vicinity of Fe–C interfaces, while fewer electrons are located on the graphene substrate. It is clearly shown that the uneven distribution of the spin charge induces the magnetic property of the system. Compared to the Fe/MG system, the much more pronounced charge density redistribution at the Fe–DG interface induces a large magnetic property, as shown in Fig. 2(b). Although the Fe atom provides more electrons to the DG substrate, the relatively weaker interaction of Fe with DG compared with MG is likely due to the bare DG being more stable than the bare MG. Because the optimized bare DG structure forms two pentagons and one octagon at the divacancy site, this quenches the dangling bonds and renders it a stable structure. Compared to the bare MG and DG sheets (12.0 and 10.0 μB), the doped Fe atom induces a change in the magnetic properties of the systems (e.g., the Fe/MG and Fe/DG system display magnetic property values of 10.0 and 13.4 μB), which is accordance with the reported results.53 For the metal-decorated graphene system, the hybridization of the metal atom d states and π states of graphene lead to their enhanced interaction. The metal dopant tends to provide more transferred electrons to graphene and induces spin charge redistribution at their interface, which can effectively regulate the electronic structure and magnetic property of graphene-based materials.
 | ||
| Fig. 2 Spin charge density maps for (a) Fe/MG and (b) Fe/DG sheets; the contour value is 0.001 e Å−3 intervals. | ||
3.2. Adsorption of CO and O2 molecules
Based on the highly stable structure of the Fe–graphene substrates, as shown in Fig. 1(a) and (b), we examined the possible adsorption sites in order to find out the most energetically favorable configurations for the reactant gases on Fe/MG or Fe/DG substrates. The corresponding adsorption energies and structural parameters for the optimized structures are listed in Table 1. It was found that the adsorption energies of individual O2 on the Fe–graphene systems are larger than those for the CO molecule. The adsorptive O2 has roughly similar configurations on the different graphene substrates, where it prefers to form two bonds with the Fe atom and where the O–O bond is parallel to the Fe–graphene surface, as shown in Fig. 3(a) and (c). Compared to the adsorbed O2 on the Fe/DG (1.80 eV), the single O2 has a relatively larger adsorption energy (1.88 eV) on Fe/MG, which is 0.33 eV more favorable than it would be at the end-on configuration. There are also more electrons (0.81 e) transferred from the Fe/MG to the adsorbed O2, which subsequently leads to elongating the O–O bond (1.40 Å). In comparison, the adsorbed O2 gains fewer electrons (0.53 e) on the Fe/DG substrate and the O–O bond length is thus shorter at 1.34 Å. | ||
| Fig. 3 Top and side views of the geometric structures for (a)–(c) O2 and (b)–(d) CO adsorbed on the Fe/MG and Fe/DG sheets, respectively. The green, black, and red balls represent Fe, C, and O atoms, respectively. | ||
As shown in Fig. 3(b)–(d), the most stable configurations of the CO molecule adsorbs on the Fe/MG and Fe/DG substrates. It was found that the end-on adsorbed CO is nearly vertical on the Fe/MG surface, with the distance between Fe and CO equal to 1.88 Å, and it has a smaller Eads (1.13 eV) than that on the Fe/DG substrate (1.77 eV). In comparison, the adsorbed CO is somewhat tilted on the Fe/DG surface, with an Fe–CO distance of 1.78 Å. Herein, we report that we found that the adsorption of O2 on the Fe–graphene systems were more stable than those of the CO molecule, since there were more transferred electrons from the bound Fe atom to the O2 molecule. Furthermore, the elongation of the O–O bond was connected to the number of transferred electrons from the Fe–graphene substrates.
The electronic structure, which fundamentally determines the physical and chemical properties of a system, is directly related to the interaction between the adsorbate and the substrate. For the adsorbed O2 and CO on Fe/MG systems, the corresponding DOS plots were investigated and are shown in Fig. 4. As shown in Fig. 4(a), the broadened PDOS of the Fe 3d states strongly hybridizes with the O2 2π*, 5σ and 1π orbitals around the EF, indicating that there are about 0.81 e transferred from Fe/MG to the adsorbed O2, which occupy the 2π* orbital of O2, resulting in elongation of the O–O bond to 1.40 Å. Besides, the hybridization between the Fe and O states induces the magnetic moment of the whole system (12 μB) due to the increase in the number of unpaired electrons, thus the spin-up and spin-down channels of the system become asymmetric.
 | ||
| Fig. 4 Spin-resolved TDOS, local DOS (LDOS), PDOS (spin-up labeled with ↑ and spin-down labeled with ↓) for (a) O2 and (b) CO on the Fe/MG sheet. The solid (dashed) curves represent the TDOS of Fe/MG without (with) O2 or CO adsorption. The dotted curves represent the PDOS of Fe 3d states with O2 or CO adsorption, and the dash dotted curves represent the LDOS of adsorbed O2 (or CO). The vertical dotted line denotes the Fermi level. | ||
The DOS plot for the CO on Fe/MG is shown in Fig. 4(b), where it can be observed that the hybridization between the PDOS of the Fe-3d states and the CO-2π* and 5σ states are near the EF. Compared to the adsorbed O2, fewer electrons are transferred from the Fe atom to the CO molecule (0.31 e), indicating a weak interaction between the CO and the Fe/MG. Besides, the CO on the Fe/MG system exhibits a magnetic property (10 μB) according to the asymmetry of the spin channels. Therefore, the magnetic properties of graphene systems can be tuned by choosing the kind of gas molecules adsorbed, which may have a bearing on some important applications in electronic and spintronic devices.
To further understand the origin of the differences in the electronic structure and magnetic properties according to the type of adsorbates, we investigated the valence charge density (or spin electron redistribution) of the adsorption of reaction gases on Fe–graphene substrates, as shown in Fig. 5. It was found that the adsorbed O2 and CO on Fe/MG induce spin charge redistribution at their interfaces. The more electrons that dominantly accumulate in the vicinity of the O2–Fe or CO–Fe interfaces, the fewer electrons are located on the graphene substrates. Compared to the CO molecule (10.0 μB), the adsorbed O2 on the Fe/MG sheet induces a more pronounced spin charge distribution and it thus exhibits a larger magnetic property (12.0 μB). Bader charge analysis showed that the Fe atoms lose about 1.44 and 1.23 e in these systems, in which some are partly transferred to the O2 and CO (0.81 and 0.31 e) and the rest are transferred to the graphene sheets, illustrating that the electrons deplete from the vicinity of the Fe dopants and that the transferred electrons can facilitate the interaction between the gas molecules and graphene substrates.
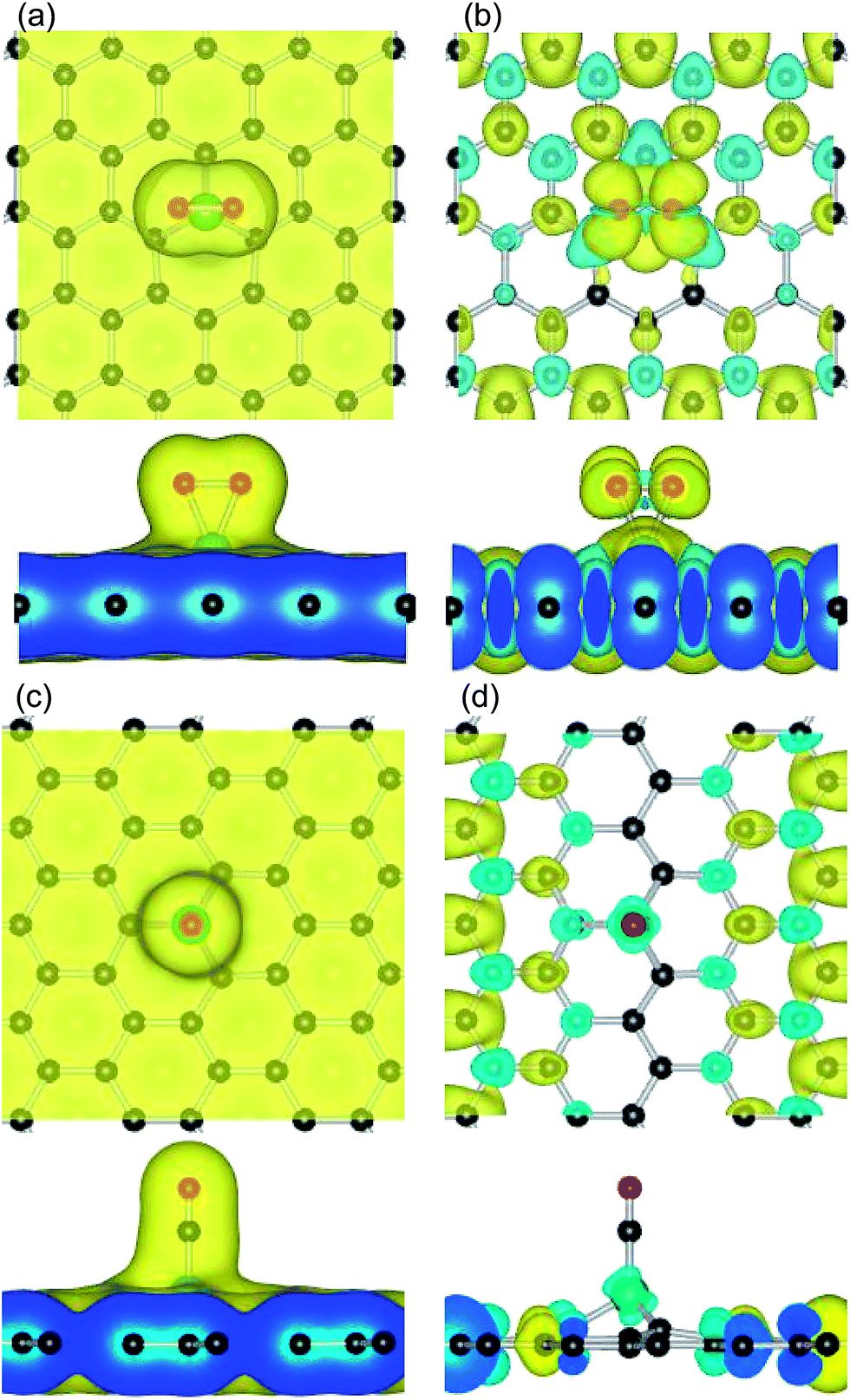 | ||
| Fig. 5 The charge distribution and spin charge density plots for (a) and (b) O2 and (c) and (d) CO adsorbed on the Fe/MG sheet. Contour lines in plots are drawn at about 0.01 (or 0.001) e Å−3 intervals. | ||
Compared with the bare Fe–graphene systems, the charge redistribution plots due to the adsorption of O2 and CO on the Fe–graphene substrates. The more pronounced charge density redistribution and the bond elongation of O2 on the Fe/MG sheet (0.81 e, 1.40 Å) compared to those on the Fe/DG sheet (0.53 e, 1.34 Å) indicate a stronger interaction of O2 on Fe/MG (1.88 eV) than that on Fe/DG (1.80 eV). Hence, the elongation of the O–O bond is correlated with the amount of electronic charge transferred to O2, i.e., the more charge is transferred to O2 from the Fe–graphene systems, the more elongated the O–O bonds become. In contrast, CO gains fewer electrons on the Fe–graphene substrates compared with the adsorbed O2 molecules, as shown in Table 1. The larger charge density accumulation between the adsorbed O2 and Fe–graphene illustrate that their interaction is stronger than that for the adsorption of CO. For the adsorbed CO on Fe–graphene, it was found that the small difference in electron gain induces a larger energy difference, indicating that CO is a more sensitive gas molecule and can be easily influenced by various conditions; or example, the Fe dopant in the Fe/MG system largely weakens the CO adsorption (1.13 eV) compared with that on the Fe/DG (1.77 eV). Hence, the interaction of Fe atoms with different graphene systems would modify the electronic structure and catalytic activity of the Fe catalyst. The losing electrons from the Fe dopants exhibit a positive charge, which can alleviate the CO adsorption and enhance the adsorptive stability of the O2 molecule, and thus the Fe–graphene sheet is more likely to be useful as a catalytic anode material for facilitating the catalytic reaction of CO oxidation.
Generally, the degree of interaction or the strength of the bonding can determine the reaction energy barriers.70 Consequently, it is desired that the interactions between the active sites of catalysts and reactive gases should be adequate. Our calculated results show that the transferred electrons from the Fe dopants can promote the interaction between reactive gases and graphene substrates. Compared to the CO molecule, O2 is viewed as an electron-acceptor molecule.67 As they gain more electrons, O2 molecules have larger adsorption energies on the Fe–graphene substrates compared to CO. The anchored Fe atom on graphene prefers to be covered by adsorbed O2 if a mixture of CO and O2 is injected as the reaction gas. Since O2 has a larger adsorption energy than that of the CO molecule, the adsorption of O2 on Fe–graphene substrates takes a higher priority and thus the ER mechanism seems to be more favorable. Besides, the coadsorption of CO and O2 molecules involve larger Eads on the Au–graphene (1.82 eV),64 Al–graphene (1.95 eV),69 Cu–graphene (3.29 eV),66 Fe/MG (2.10 eV) and Fe/DG (2.60 eV) sheets than that for the individual CO or O2 molecules, as shown in Table S1.† Although the adsorption energy difference between CO and O2 on the Fe/MG is relatively large, the lower coadsorption energy of the O2 and CO molecules indicates that there is a certain probability of having O2 and CO coadsorbed on the Fe atom. Therefore, it is worth exploring the catalytic activity of Fe–graphene sheets through comparing different reaction mechanisms for CO oxidation.
3.3. Interaction mechanism between the adsorbed O2 and CO
First, several coadsorption configurations of CO and O2 molecules on Fe/MG through the LH mechanism were tested. The most stable coadsorption configuration was viewed as an IS, where the CO and O2 are tilted and parallel to the Fe/MG surface, respectively. The FS consists of a physisorbed CO2 molecule and a chemisorbed Oads. For CO oxidation, the local configurations between the reactants on the Fe/MG surface at each state along the MEP are displayed in Fig. 6(a), and the corresponding structural parameters for IS, TS, MS, and FS are shown in Table 2(a).
 | ||
| Fig. 6 The minimum energy profiles and configurations of different states for the CO oxidation reaction on the Fe/MG sheet, including (a) CO + O2 reaction by the LH mechanism, (b) CO + O2 = CO2 + Oads and (c) CO + Oads reactions by the ER mechanism. The red, green, and black balls represent O, Fe, and C atoms, respectively. | ||
| (a) | |||||
|---|---|---|---|---|---|
| Distance (Å) | IS | TS1 | MS | TS2 | FS |
| dC–O | 1.15 | 1.16 | 1.20 | 1.17 | 1.18 |
| dC–Fe | 1.96 | 2.03 | 2.07 | 2.65 | 3.79 |
| dC–O1 | 2.61 | 1.95 | 1.37 | 1.17 | 1.17 |
| dO1–Fe | 1.99 | 2.46 | 2.56 | 2.90 | 4.39 |
| dO2–Fe | 2.00 | 1.91 | 1.88 | 1.65 | 1.62 |
| dO1–O2 | 1.33 | 1.36 | 1.48 | 2.12 | 2.99 |
| (b) | |||
|---|---|---|---|
| Distance (Å) | IS | TS | FS |
| dO1–O2 | 1.39 | 1.34 | 2.79 |
| dC–O1 | 3.19 | 2.29 | 1.16 |
| dO1–Fe | 1.86 | 2.67 | 3.52 |
| (c) | |||
|---|---|---|---|
| dO2–Fe | 1.62 | 1.68 | 1.84 |
| dO2–C | 2.72 | 1.75 | 1.16 |
| dC–O | 1.14 | 1.16 | 1.15 |
Once the coadsorption of CO and O2 takes place on Fe/MG, the distance between O2 and CO is about 2.38 Å, while the Fe–CO and Fe–O2 distances are 1.93 and 2.04 Å, respectively. To proceed, the O2 molecule turns around so that one of its oxygen atoms now approaches the CO molecule, while another oxygen atom is anchored at the Fe atom. Passing over the TS1, the OOCO complex (MS) is formed, and the corresponding energy barrier (Ebar) along the MEP is estimated to be 0.29 eV. In this reaction process, the bond length of O2 (dO1–O2) is gradually elongated from 1.34 to 1.48 Å. The reaction continuously proceeds from MS to FS through TS2 without any energy barrier, where the O1–O2 bond is broken and a CO2 molecule is formed, leaving an atomic O2 to adsorb at the Fe atom, which is expected to be active and can be used for the CO oxidation reaction. According to the reported result by Chen,91 we checked the possible reactions for CO oxidation through the ER mechanism without any intermediate state (i.e., CO + O2 = CO2 + Oads), as shown in Fig. 6(b). The first physisorbed CO above O2 preadsorbed on the Fe/MG sheet was selected as the IS, and the distance between CO and O2 was 3.19 Å. As shown in Table 2(b), the migration of the CO molecule involves moving toward the preadsorbed O2 molecule and the distance changes from 3.19 to 1.16 Å, and then CO2 is generated, where the calculated energy barrier is 0.56 eV in this process. After desorbing of the CO2 molecule, we further checked the oxidation process of a second CO molecule reacting with the atomic O2 to produce CO2. As shown in Fig. 6(c), the configuration with a CO molecule more than 2.72 Å away from the preadsorbed Oads (Eads, 5.45 eV) on the Fe atom is viewed as the IS. The adsorption configuration of CO2 on the Fe/MG sheet is considered as the FS. When the carbon atom of CO approaches the atomic O2, a TS with a CO–Oads distance of 1.75 Å is formed, as shown in Table 2(c). It was found that the CO oxidation through the first ER reaction (CO + O2 = CO2 + Oads) has a relatively larger energy barrier (0.56 eV) than that of the second one (CO + Oads = CO2, 0.38 eV), which are both higher than for the first step (TS1, 0.29 eV) through the LH reaction.
As an important reference, the physisorbed CO molecule directly reacts with the preadsorbed O2 through the ER mechanism through an intermediate state (i.e., CO + O2 → CO3), with the corresponding atomic configurations of each state along the MEP displayed in Fig. 7 and Table 3. At first, the configuration of the physisorbed CO above O2 preadsorbed on the Fe/MG sheet was selected as an IS. When approaching the activated O2, one CO molecule can be inserted into the O–O bond to form a carbonate-like CO3 complex (MS) with a small energy barrier of 0.32 eV (TS1). Then, the reaction can proceed with the dissociating of CO3 into CO2 and leaving an Oads atom (FS). The energy barrier (TS2) for this process was estimated to 0.72 eV, which is quite similar to the case of CO oxidation on Mo–graphene92 and Pt-anchored graphene oxide.93 However, it was found that the formed CO3 is more stable than the final products (CO2 and Oads), since the reversible reaction (CO2 + Oads → CO3) has a relatively small energy barrier (0.41 eV), as shown in Fig. 7(a), and consequently the more stable CO3 is an energetically unfavorable reaction for generating CO2. Furthermore, the possible reaction pathway after the formation of CO3 is to react with another CO molecule (IS) to produce two CO2 molecules (FS) through the TS, as shown in Fig. 7(b). In this reaction (CO3 + CO → 2CO2), the corresponding energy barrier (0.61 eV) is smaller than the direct dissociation energy of the CO3 complex (0.72 eV), indicating that the presence of a CO molecule can promote the dissociation of a CO3 complex on the Fe/MG surface.
 | ||
| Fig. 7 The minimum energy profiles and configurations of different states for the CO oxidation reaction on the Fe/MG sheet, including (a) CO + O2 and (b) CO3 + CO reactions by the ER reactions. The red, green, and black balls represent O, Fe, and C atoms, respectively. | ||
| (a) | |||||
|---|---|---|---|---|---|
| Distance (Å) | IS | TS1 | MS | TS2 | FS |
| dC–O | 1.14 | 1.14 | 1.21 | 1.18 | 1.17 |
| dC–O1 | 3.00 | 2.42 | 1.35 | 1.19 | 1.17 |
| dC–O2 | 3.00 | 2.42 | 1.35 | 2.07 | 2.56 |
| dO1–Fe | 1.87 | 1.85 | 1.91 | 2.70 | 3.35 |
| dO2–Fe | 1.87 | 1.85 | 1.91 | 1.65 | 1.62 |
| dO1–O2 | 1.40 | 1.43 | 2.18 | 2.52 | 2.87 |
| (b) | |||
|---|---|---|---|
| Distance (Å) | IS | TS | FS |
| dO2–C | 1.36 | 2.21 | 2.80 |
| dO2–C2 | 3.01 | 3.19 | 1.18 |
| dO2–Fe | 1.89 | 1.63 | 2.59 |
After the dissociation reaction of an O2 molecule on Fe/MG, we further checked whether the dissociated atomic Oads is active for CO oxidation through ER reactions, as shown in Fig. 8. In these reaction processes, the O–O bond is elongated from 1.40 to 3.06 Å and the formed two O atoms are anchored at the top site of the carbon atoms, and the corresponding energy barrier is estimated to be 1.01 eV, as shown in Fig. 8(a). As shown in Fig. 8(b), the CO oxidation reactions on the Fe/MG surface were investigated, and these include a two-step process for the interaction between two O atoms and two CO molecules. First, the configuration with preadsorbed Oads and a subsequent CO near the Fe is viewed as the IS, where the CO–O distance is 3.11 Å. It was found that the CO reaches and reacts with the Oads through an ER reaction, with an Ebar of 0.67 eV. Second, the physisorbed CO molecule is close to the binding O atom, where the corresponding distance CO–O is 3.15 Å. When the carbon atom of CO approaches the O atom, it generates a second CO2 with an Ebar of 0.79 eV. In the sequential reactions, it was found that the dissociative adsorption of an O2 molecule (1.01 eV) is more difficult than that of the formed CO2 molecules (0.67 and 0.79 eV), yet these energy barriers are much larger than that on the top site of the Fe atom (CO + O → CO2, 0.38 eV). This indicates that the dissociative adsorption of O2 as a starting step is an energetically unfavorable reaction for CO oxidation. Based on the above discussions, the coadsorption of CO and O2 through the LH reaction (0.29 eV) has a much smaller Ebar than those of the ER reactions. Hence, the LH reaction as a starting step is the more favorable process on the Fe/MG surface.
 | ||
| Fig. 8 The minimum energy profiles and configurations of different states for the CO oxidation reaction on the Fe/MG sheet, including (a) the dissociative adsorption of an O2 molecule and (b) 2CO + 2Oads reactions by the ER mechanism. The red, green, and black balls represent O, Fe, and C atoms, respectively. | ||
For the CO oxidation reactions on the Fe–graphene substrates, the coadsorption of CO and O2 as the starting step has a much lower energy barrier on the Fe/MG sheet (0.29 eV), while, the second step for CO oxidation through the ER reaction involves a relatively large energy (0.38 eV). Thus, the complete CO oxidation reactions on the Fe/MG substrate include a two-step process: the LH reaction as the starting step (CO + O2 → OOCO → CO2 + Oads), followed by the ER reaction (CO + Oads → CO2). Compared with the LH reactions, the formation of a CO2 molecule through the ER reaction involves a larger energy barrier, and so it can be viewed as the rate limiting step. Furthermore, these energy barriers are low enough (<0.4 eV) for the catalytic reaction to proceed rapidly at low temperature.64 For the Fe/DG sheet, it was found that the CO oxidation processes through the ER reactions have smaller energy barriers (<0.7 eV) than those of the LH reaction, illustrating that the ER reaction as a starting state is a favorable mechanism. In the ER reactions, the oxidation process on the Fe/DG (CO + Oads → CO2, 0.68 eV) has a larger energy barrier than that of the other processes (0.30 eV, 0.47 eV, and 0.66 eV), and so it can be viewed as the rate limiting step. These results indicate that the reaction efficiency and energy barriers can be controlled by choosing different substrates or reaction mechanisms. For the Fe/MG sheet, the large energy difference and the low coadsorption energy of the reaction gases through LH mechanism may promote the catalytic reaction of CO oxidation. In comparison, the larger coadsorption energy of CO and O2 on the Fe/DG sheet need to overcome a higher activation energy barrier, thus the LH mechanism as a starting step is energetically unfavorable. So, the CO oxidation reaction on the Fe/DG sheet through the ER mechanism is more possible or proceeds with greater ease. Compared with the ER reaction, the LH reactions on the Fe/MG sheet have smaller energy barriers, which ensure high efficiency in the catalytic reaction of CO oxidation.
In light of the aforementioned discussion, it can be concluded that the sequential reactions of CO oxidation through LH reactions have low enough energy barriers on the Fe/MG sheet (<0.4 eV), while the ER reaction as an IS on the Fe/DG sheet is an energetically more favorable process. These results indicate that the different vacancies in graphene sheets can regulate the stability and catalytic activity of the metal Fe dopant. Compared with the LH reaction on Cu– (0.54 eV),66 Pd– (0.54 eV),68 Pt– (0.59 eV),67 Co– (0.42 eV),94 Al– (0.32 eV),69 Au–graphene (0.31 eV),64 and Fe/DG (0.91 eV), the CO oxidation reaction on the Fe/MG sheet has a smaller energy barrier (0.29 eV). The sequential processes of CO oxidation on the Fe/MG sheet are more likely to proceed rapidly in practical reactions because of the low activation barriers involved. Therefore, single-atom Fe catalyst-embedded graphene substrates have a high activity for the catalytic oxidation CO reaction, which provides a valuable reference to fabricate functionalized graphene-based devices with low cost and high catalytic efficiency.
4. Conclusions
In summary, we carried out a comprehensive study of the structural stability and CO oxidation activity of single-atom Fe catalyst-embedded MG and DG sheets by means of DFT computations. It was found that the Fe dopants in MG and DG sheets introduce electronic doping, which can significantly regulate the electronic structure and magnetic property of the systems, as well as the strength of the interaction between the reactants and Fe–graphene. The individual O2 molecule can be strongly bonded and activated on the Fe–graphene sheets with larger adsorption energies than that of a CO molecule. In addition, the catalytic reactions of CO oxidation were investigated and compared through comparing the LH and ER mechanisms. As a result, the CO oxidation reactions were found to be more likely to proceed rapidly on the Fe/MG sheet through the LH reaction (CO + O2 → OOCO → CO2 + Oads, 0.29 eV), followed by the ER reaction (CO + Oads → CO2, 0.38 eV), because these catalytic processes have low enough energy barriers. In comparison, the ER reaction as the starting step is a favorable mechanism on the Fe/DG sheet. Our results demonstrate that the Fe–graphene sheets are promising anode materials for CO oxidation and can proceed at low temperature, and this report provides a comprehensive understanding of graphene–metal composite materials for the fields of catalysis and gas sensing.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. U1404109, 11504334, 51401078 and U1504108), the Application Foundation and Advanced Technology Research Program of Henan Province (Grant No. 152300410167).References
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650–3653 CrossRef CAS.
- M. Ackermann, T. Pedersen, B. Hendriksen, O. Robach, S. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505 CrossRef CAS PubMed.
- C. Zhang and P. Hu, J. Am. Chem. Soc., 2001, 123, 1166–1172 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- S. H. Oh and G. B. Hoflund, J. Catal., 2007, 245, 35–44 CrossRef CAS.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Z. P. Liu, X. Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef PubMed.
- X. Q. Gong, Z. P. Liu, R. Raval and P. Hu, J. Am. Chem. Soc., 2004, 126, 8–9 CrossRef CAS PubMed.
- L. Molina and B. Hammer, Phys. Rev. Lett., 2003, 90, 206102 CrossRef CAS PubMed.
- I. Nakai, H. Kondoh, K. Amemiya, M. Nagasaka, A. Nambu, T. Shimada and T. Ohta, J. Chem. Phys., 2004, 121, 5035 CrossRef CAS PubMed.
- M. Nagasaka, H. Kondoh, I. Nakai and T. Ohta, J. Chem. Phys., 2007, 126, 044704 CrossRef PubMed.
- M. Chen, Y. Cai, Z. Yan, K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS.
- X. Liu, A. Wang, X. Wang, C. Y. Mou and T. Zhang, Chem. Commun., 2008, 27, 3187–3189 RSC.
- S. N. Rashkeev, A. R. Lupini, S. H. Overbury, S. J. Pennycook and S. T. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 035438 CrossRef.
- C. Chang, C. Cheng and C. Wei, J. Chem. Phys., 2008, 128, 124710 CrossRef CAS PubMed.
- N. Lopez and J. K. Nørskov, J. Am. Chem. Soc., 2002, 124, 11262–11263 CrossRef CAS PubMed.
- H. Falsig, B. Hvolbak, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Norskov, Angew. Chem., 2008, 120, 4913–4917 CrossRef.
- B. Kalita and R. C. Deka, J. Am. Chem. Soc., 2009, 131, 13252–13254 CrossRef CAS PubMed.
- H. Kim, S. Han, J. Ryu and H. Lee, J. Phys. Chem. C, 2010, 114, 3156–3160 CAS.
- S. Dobrin, Phys. Chem. Chem. Phys., 2012, 14, 12122–12129 RSC.
- B. H. Morrow, D. E. Resasco, A. Striolo and M. B. Nardelli, J. Phys. Chem. C, 2011, 115, 5637–5647 CAS.
- W. Liu, Y. Zhao, R. Zhang, Y. Li, E. J. Lavernia and Q. Jiang, ChemPhysChem, 2009, 10, 3295–3302 CrossRef CAS PubMed.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- D. Ma, Z. Wang, H. Cui, J. Zeng, C. He and Z. Lu, Sens. Actuators, B, 2016, 224, 372–380 CrossRef CAS.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- D. Ma, Q. Wang, T. Li, Z. Tang, G. Yang, C. He and Z. Lu, J. Mater. Chem. C, 2015, 3, 9964–9972 RSC.
- C. Du, H. Lin, B. Lin, Z. Ma, T. Hou, J. Tang and Y. Li, J. Mater. Chem. A, 2015, 3, 23113–23119 CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- A. Geim and K. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- K. Novoselov, A. Geim, S. Morozov, D. Jiang, M. Katsnelson, I. Grigorieva, S. Dubonos and A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung and E. Tutuc, Science, 2009, 324, 1312 CrossRef CAS PubMed.
- Y. Tang, Z. Yang and X. Dai, J. Nanopart. Res., 2012, 14, 844 CrossRef.
- Y. Tang, Z. Lu, W. Chen, W. Li and X. Dai, Phys. Chem. Chem. Phys., 2015, 17, 11598–11608 RSC.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- R. Siburian and J. Nakamura, J. Phys. Chem. C, 2012, 116, 22947–22953 CAS.
- E. Yoo, T. Okada, T. Akita, M. Kohyama, I. Honma and J. Nakamura, J. Power Sources, 2011, 196, 110–115 CrossRef CAS.
- G. Chen, S. J. Li, Y. Su, V. Wang, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. C, 2011, 115, 20168–20174 CAS.
- B. Seger and P. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CAS.
- N. Cuong, A. Sugiyama, A. Fujiwara, T. Mitani and D. Chi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235417 CrossRef.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC.
- A. Kasry, M. A. Kuroda, G. J. Martyna, G. S. Tulevski and A. A. Bol, ACS Nano, 2010, 4, 3839–3844 CrossRef CAS PubMed.
- M. N. Groves, C. Malardier-Jugroot and M. Jugroot, J. Phys. Chem. C, 2012, 116, 10548–10556 CAS.
- F. Li, J. Zhao and Z. Chen, J. Phys. Chem. C, 2012, 116, 2507–2514 CAS.
- M. N. Groves, A. S. W. Chan, C. Malardier-Jugroot and M. Jugroot, Chem. Phys. Lett., 2009, 481, 214–219 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, G. Xu, T. Wang, D. Ma, Z. Yang and L. Yang, Phys. Chem. Chem. Phys., 2015, 17, 20006–20013 RSC.
- Y. Tang, Z. Yang, X. Dai, D. Ma and Z. Fu, J. Phys. Chem. C, 2013, 117, 5258–5268 CAS.
- A. Krasheninnikov, P. Lehtinen, A. Foster, P. Pyykkö and R. Nieminen, Phys. Rev. Lett., 2009, 102, 126807 CrossRef CAS PubMed.
- O. Cretu, A. V. Krasheninnikov, J. A. Rodríguez-Manzo, L. Sun, R. M. Nieminen and F. Banhart, Phys. Rev. Lett., 2010, 105, 196102 CrossRef PubMed.
- J. Rodriguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef CAS PubMed.
- Y. N. Tang, Z. X. Yang and X. Q. Dai, J. Chem. Phys., 2011, 135, 224704 CrossRef PubMed.
- E. J. G. Santos, D. Sanchez-Portal and A. Ayuela, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125433 CrossRef.
- E. J. G. Santos, A. Ayuela and D. Sánchez-Portal, New J. Phys., 2010, 12, 053012 CrossRef.
- Y. Mao and J. Zhong, Nanotechnology, 2008, 19, 205708 CrossRef PubMed.
- A. Ambrosi, S. Y. Chee, B. Khezri, R. D. Webster, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2012, 51, 500–503 CrossRef CAS PubMed.
- J. A. Rodriguez-Manzo and F. Banhart, Nano Lett., 2009, 9, 2285–2289 CrossRef CAS PubMed.
- Y. J. Gan, L. T. Sun and F. Banhart, Small, 2008, 4, 587–591 CrossRef CAS PubMed.
- H. Wang, Q. Wang, Y. Cheng, K. Li, Y. Yao, Q. Zhang, C. Dong, P. Wang, U. Schwingenschlögl, W. Yang and X. X. Zhang, Nano Lett., 2012, 12, 141–144 CrossRef CAS PubMed.
- Y. Lu, M. Zhou, C. Zhang and Y. Feng, J. Phys. Chem. C, 2009, 113, 20156–20160 CAS.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CAS.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678–3683 CAS.
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566–16572 RSC.
- T.-T. Jia, C.-H. Lu, Y.-F. Zhang and W.-K. Chen, J. Nanopart. Res., 2014, 16, 1–11 Search PubMed.
- Q. G. Jiang, Z. M. Ao, S. Li and Z. Wen, RSC Adv., 2014, 4, 20290–20296 RSC.
- H. Liu, B. Wang, M. Fan, N. Henson, Y. Zhang, B. F. Towler and H. G. Harris, Fuel, 2013, 113, 712–718 CrossRef CAS.
- Y. Tang, X. Dai, Z. Yang, Z. Liu, L. Pan, D. Ma and Z. Lu, Carbon, 2014, 71, 139–149 CrossRef CAS.
- Z. Lu, S. Li, P. Lv, C. He, D. Ma and Z. Yang, Appl. Surf. Sci., 2016, 360, 1–7 CrossRef CAS.
- L. Xin, S. Yanhui, D. Ting, M. Changong and H. Yu, Phys. Chem. Chem. Phys., 2014, 16, 23584–23593 RSC.
- M. Ugeda, D. Fernández-Torre, I. Brihuega, P. Pou, A. Martínez-Galera, R. Pérez and J. Gómez-Rodríguez, Phys. Rev. Lett., 2011, 107, 116803 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- M. T. Lusk and L. D. Carr, Phys. Rev. Lett., 2008, 100, 175503 CrossRef PubMed.
- A. Barinov, H. Ustunel, S. Fabris, L. Gregoratti, L. Aballe, P. Dudin, S. Baroni and M. Kiskinova, Phys. Rev. Lett., 2007, 99, 46803 CrossRef PubMed.
- A. Hashimoto, K. Suenaga, A. Gloter, K. Urita and S. Iijima, Nature, 2004, 430, 870–873 CrossRef CAS PubMed.
- D. H. Lim and J. Wilcox, J. Phys. Chem. C, 2011, 115, 22742–22747 CAS.
- S. Kattel, P. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 8161–8166 CAS.
- A. W. Robertson, B. Montanari, K. He, J. Kim, C. S. Allen, Y. A. Wu, J. Olivier, J. Neethling, N. Harrison and A. I. Kirkland, Nano Lett., 2013, 13, 1468–1475 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Carlsson and M. Scheffler, Phys. Rev. Lett., 2006, 96, 46806 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- T. Zhu, J. Li and S. Yip, Phys. Rev. Lett., 2004, 93, 25503 CrossRef PubMed.
- Z. W. Chen, J. M. Yan, W. T. Zheng and Q. Jiang, Sci. Rep., 2015, 5, 11230 CrossRef CAS PubMed.
- Y. Tang, L. Pan, W. Chen, C. Li, Z. Shen and X. Dai, Appl. Phys. A, 2015, 119, 475–485 CrossRef CAS.
- Y. Tang, X. Dai, Z. Yang, L. Pan, W. Chen, D. Ma and Z. Lu, Phys. Chem. Chem. Phys., 2014, 16, 7887–7895 RSC.
- Y. Tang, D. Ma, W. Chen and X. Dai, Sens. Actuators, B, 2015, 211, 227–234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14476d |
| This journal is © The Royal Society of Chemistry 2016 |