
Novel one-pot synthesis and sensitisation of new BiOCl–Bi2S3 nanostructures from DES medium displaying high photocatalytic activity†
 *a
*a
Abstract
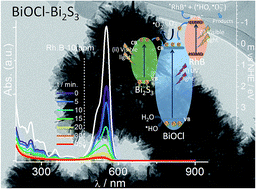
A novel route to synthesise Bi2S3-sensitised BiOCl nanoparticles from deep eutectic solvent medium at room temperature by a one-pot approach is reported. The influence of the temperature, sulphur source, concentration of reactants and presence of water, on the morphological, structural and microstructural, optical and photocatalytic properties of the synthesised nanoparticles is analysed and discussed. Stable and crystalline BiOCl hybrid structures with shapes from sheet-like to flower-like hierarchical aggregates and (001) and (110) dominant crystallographic orientation were obtained. The sensitisation of BiOCl with Bi2S3 was successfully achieved in situ during synthesis by an ion-exchange process and the relative proportion of the components (BiOCl and Bi2S3) was controlled by the Bi : S ratio in the synthesis medium and by the sulphur precursor. The sensitiser nanomaterial (Bi2S3) extends the BiOCl photoactive region to the visible range. Also it favours charge separation and reducing the electron/hole pair recombination and therefore increasing the photocatalytic performance. The prepared composite materials show high ability to adsorb rhodamine B cationic dye and the complete photocatalytic degradation was achieved within 45 min (75 mg per g of catalyst).
 Please wait while we load your content...
Please wait while we load your content...

