DOI:
10.1039/C6RA14408J
(Paper)
RSC Adv., 2016,
6, 80630-80639
A property-performance correlation and mass transfer study of As(V) adsorption on three mesoporous aluminas†
Received
3rd June 2016
, Accepted 16th August 2016
First published on 16th August 2016
Abstract
High adsorption capacity and quick adsorption kinetics are necessary for excellent adsorbents. Three different mesoporous aluminas (MA) were extensively characterized to determine their key structural properties that account for their good adsorption capacities and fast kinetics for sequestering As(V) from water. MA1 showed the largest As(V) adsorption capacity of 175.7 mg g−1 among the three. No direct relationship was observed between the mesoporous pore properties and the adsorption capacities of these mesoporous aluminas. The extraordinarily large adsorption capacity of MA1 was correlated to its strongly disordered state confirmed by a pair distribution function (PDF) analysis, its large amorphous content fraction measured by selective chemical extraction (SCE), its high Al–OH surface site density determined by a surface titration method, and its Al–O coordination environment (AlO4 and AlO5) identified by 27Al NMR. These results led to the realization that extensive structural disorder, large amorphous content and high surface site density are preferred in future adsorbent preparations. Besides its excellent adsorption capacity, MA1 also had the fastest adsorption kinetics. All As(V) uptake kinetics data were modeled using the homogeneous surface diffusion model (HSDM). The intraparticle surface diffusion coefficients (Ds) were numerically determined. The fast As(V) uptake kinetics of MA1 were explained by it having the smallest particle size from the HSDM calculations. Decreasing solution pH significantly improved the As(V) treatment efficiencies. Maximum sorption occurred at pH below 5.0. MA1 effectively decreased As(V) concentrations in spiked well water to well below the WHO's maximum level of As(V) in drinking water, indicating its great potential as a practical adsorbent candidate.
1. Introduction
Arsenic (As) is a ubiquitous toxic metalloid widely occurring in the environment due to natural geological processes (e.g. weathering and volcanic eruption) and anthropogenic activities.1 In aquatic environments, inorganic (As(V) and As(III)) species predominate over organic species. H2AsO4− and HAsO42− are the most stable As(V) species over a typical pH range (4–9), while uncharged pyramidal arsenous acid, As(OH)3 is the dominant As(III) species.2
Arsenic contaminated drinking water remains a major worldwide health concern in many areas, especially in Bangladesh, India, USA, China, Mexico, Chile, etc.3 Long-term arsenic ingestion can cause lung, bladder, liver, kidney, and other cancers.4 A 10 μg L−1 arsenic regulation limit in drinking water has been set by many agencies and countries including WHO, US EPA, Health Canada, the Ministry of Health, China, EU, and Japan.3
A variety of arsenic removal technologies from drinking water exist, such as coagulation–filtration,5 oxidation–precipitation,6 adsorption,7 ion exchange,8 and membrane separation.9 As one of the most promising technologies for rural and small community water supplies, adsorption is considered to have low cost, design simplicity, easy operation and little risk.10 An efficient adsorbent is the core of this technology. Metal oxide/hydroxides of iron, aluminium, manganese, zirconium and titanium are widely investigated and commercially available adsorbents, with satisfactory performances, costs and health profiles.11,12 Activated alumina (AA), an aluminium oxide/hydroxide, has a relatively long history of practical arsenic removal from water. It was listed as one of the best available technologies (BAT) by the US EPA.13 Mesoporous alumina has attracted growing interest because of its tunable pore size and size distribution, large surface area and pore volume, and high thermal and mechanical stability.14 It is increasingly used for mitigation and remediation of multiple contaminants in water. These include hazardous anions (e.g. AsO43−, F−),15–17 heavy metals,18 radioactive waste,19 organic dyes,20 and rare earth elements.19 Mesoporous alumina and functionalized mesoporous aluminas, e.g., mesoporous γ-Al2O3 nanospheres,21 mesoporous silica hybrid alumina,22 copper oxide incorporated onto mesoporous alumina,23 and manganese oxide coated alumina24 have been evaluated as arsenic adsorbents. Most of these are superior to conventional activated alumina and many other widely used commercial adsorbents.24
Mesoporosity, high surface area, and large pore volume are claimed to be responsible for enhanced adsorption ability.25,26 No correlations between these properties and the adsorption capacity were also observed for alumina-containing adsorbents in some work.24,27 A thorough correlation of these three surface and bulk properties with the adsorption capacity and kinetics is necessary to design better adsorbents. To help further understand these unknowns, four fundamental properties including local atomic arrangement, Al–O coordination species (e.g. AlO4, AlO5 and AlO6), Al hydroxyl surface site density, and the amorphous content fractions were characterized in this paper on three different mesoporous aluminas. These properties were then correlated with the adsorption capacity differences among these three aluminas. In addition, the effect of adsorbent particle sizes on As(V) uptake kinetics were considered. A powerful tool for characterizing atomic arrangement, PDF, and the kinetics model, HSDM, are introduced below to help further understand these unknowns.
1.1. Pair distribution function (PDF)
Atomic pair distribution function (PDF) analysis provides a direct measure of the probability of finding an atom surrounding a central atom at a given radial distance. The pair distribution function (PDF) can be determined from Mo- or Ag-target X-ray, synchrotron X-ray, or neutron diffraction data.28 The PDF method provides a powerful tool to investigate short, medium or long-range atomic order. It can be used to characterize crystalline, amorphous, and glassy solids as well as liquids. PDF has long been used in the materials science and pharmaceutical communities.29
The pair distribution function, G(r), produced by Fourier transforming the normalized total scattering of X-ray diffraction, is defined as follows,
where
ρ(
r) is the microscopic pair density,
ρ0 is the average number density,
S(
Q) is the total scattering structure function, and
Q is the magnitude of the scattering vector.
Q is given by
Q = 4π
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
sin
θ/
λ with
θ being the scattering angle and
λ is the wavelength of the radiation. The PDF was extracted from X-ray powder scattering data by using the PDFgetX3 software.
30
1.2. The homogeneous surface diffusion model (HSDM)
The homogeneous surface diffusion model (HSDM) has been extensively used to describe the kinetics of multiple adsorbate adsorption on porous media.31 The adsorption process in this model was summarized as three steps: (1) external mass transfer, where adsorbate is transferred from the bulk of the solution to the adsorbent's external surface, (2) surface diffusion along the particle inner surface and (3) adsorption on the active sites.32 Since the resistance of the external film to mass transfer is negligible in comparison to the intraparticle mass transfer resistance in a well-stirred or rigorously shaken batch reactor,33 this resistance can be neglected. Thus the model was simplified to a Fick's second law-controlled diffusion. Then the homogeneous 1-D surface diffusion model in spherical coordinates is described by eqn (1).34,35| |
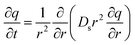 | (1) |
The initial and boundary conditions are given as,
| |
 | (2) |
| | |
q = qe, r = Rp, t = infinity
| (4) |
The average adsorbate concentration inside the adsorbent is,
| |
 | (5) |
The partial differential eqn (1) was solved numerically using the FlexPDE 6.37 student version software.36
The main objectives of this study were (1) to evaluate and compare the As(V) removal performances of the three mesoporous aluminas, (2) to unearth differences in the As(V) adsorption onto the three mesoporous aluminas by investigating the connection between the surface and bulk properties and adsorption ability using the powerful techniques of pair distribution function (PDF), selective chemical extraction (SCE), surface titration, 27Al solid-state NMR, and (3) to both model and understand the intraparticle mass transfer of As(V) on mesoporous aluminas.
2. Materials and methods
2.1. Materials
All chemicals were analytical grade except for reagent grade KBH4 and HCl, which were used for As analysis. As(V) standard solutions (200 mg L−1) were ordered from National Research Center for Certified Reference Materials, China. As(V) stock solutions (1000 mg L−1) were prepared by dissolving Na3AsO4·12H2O in deionized water. Solutions containing As(V) were freshly prepared by diluting the stock solution with distilled water.
Three mesoporous aluminas were prepared using facile methods, and the detailed procedures are provided in the ESI (S1).†
2.2. Adsorbents characterization
X-ray diffraction patterns were collected using a D8 Advance diffractometer (Bruker, German) with Cu Kα (λ = 1.54178 Å) and Mo Kα (λ = 0.7093 Å) sources, respectively. Samples were scanned at a speed of 2° min−1 from 10° to 80°, at 40 kV and 40 mA. Adsorbent surface areas, pore volumes and pore size distributions were calculated by applying the BET equation on the nitrogen adsorption data collected on a Micromeritics ASAP2000 surface area analyzer (Norcross, USA) with a degassing temperature of 230 °C applied for 6 hours. The surface morphologies were observed using a H800 high resolution transmission electron microscope (Hitachi, Japan). 27Al solid state NMR spectra were collected on an Advance II spectrometer (Bruker, Germany) at 400 MHz.
The detailed procedures used for selective chemical extraction (S2) to measure the amorphous content fractions and surface titration (S3) to determine the densities of surface hydroxyl sites are provided in the ESI.†
2.3. Batch adsorption experiments
As(V) stock solution and deionized water were added into a series of 2000 mL glass conical flasks to give a total volume of 1000 mL in each. Then, 0.3 g adsorbent was added into each flask. The pH of each suspension was adjusted and maintained at 5.0 ± 0.1 throughout the experiments. The mixtures were stirred at 130 rpm, and maintained at 25 ± 1 °C. Approximately 4 mL aliquots were taken from the suspension at predetermined intervals. These aliquots were immediately filtered through a 0.45 μm membrane, and then the residual As was analyzed. Experimental As(V) adsorption isotherms on the adsorbents were obtained at 25 ± 1 °C with initial As(V) concentrations ranging from 6 to 70 mg L−1, respectively, on a 0.3 g L−1 adsorbent dose at a pH of 5.0 ± 0.1. The effect of solution pH on As(V) removal was analyzed in 250 mL glass bottles containing 100 mL of As solution with pre-selected concentrations and a 0.3 g L−1 adsorbent dose. The pH was adjusted and maintained at a specified value in the range of 3 to 10 during a 24 h experimental period. At the beginning, pH was manually adjusted every 1 h, and then after 12 h, pH was adjusted every 2 h. After shaking for 24 h at 25 ± 1 °C, the residual As in solution was analyzed.
2.4. Analytical methods
The residual As concentrations were analyzed using a hydride generation-atom fluorescence spectrometer (HG-AFS, AF-9600, Beijing Kechuang Haiguang Instrument Corporation, China) using published methods.37
3. Results and discussion
3.1. Adsorbent characterization
HR-TEM images of the three mesoporous aluminas are shown in Fig. 1(a–c). Fig. 1 shows (a) a random aggregation of nanoparticles for MA1, (b) wormhole-like morphology for MA2, and (c) fiber- and slab-like morphologies for MA3, respectively.
 |
| | Fig. 1 TEM images of (a) MA1 (b) MA2 and (c) MA3. | |
The N2 adsorption–desorption isotherms and pore size distribution curves of the three samples are shown in Fig. 2. All three samples showed typical mesoporous features. MA1, MA2 and MA3 have specific surface areas of 166.7 m2 g−1, 543.4 m2 g−1 and 200.8 m2 g−1, respectively. Other physicochemical properties are summarized in Table 1. All three N2 adsorption–desorption isotherms were classified as type IV indicating a mesoporous nature. MA1 and MA2 (Fig. 2a and b) presented two H1 type hysteresis loops revealing a narrow pore size distribution, while MA3 exhibited a H3 type hysteresis loop revealing a broad pore size distribution (Fig. 2c).
 |
| | Fig. 2 N2 adsorption–desorption isotherms and pore size distributions (inset plots) for (a) MA1 (b) MA2 and (c) MA3. | |
Table 1 The properties of MA adsorbents
| Adsorbent |
MA1 |
MA2 |
MA3 |
| Surface area (m2 g−1) |
166.7 |
543.4 |
200.8 |
| Pore size (nm) |
5.612 |
4.221 |
7.032 |
| Average particle diameter df (μm) |
42 |
134 |
245 |
| Pore volume Vp (cm3 g−1) |
0.241 |
0.586 |
0.399 |
| Apparent density ρp (g cm−3) |
1.314 |
0.696 |
1.051 |
| Void fraction εp |
0.277 |
0.597 |
0.516 |
| Solid density ρs (g cm−3) |
1.818 |
1.726 |
2.171 |
The XRD patterns from the Cu Kα radiation sources are shown in Fig. 3. MA1 and MA2 present weak and broad peaks presenting an amorphous nature. Mesoporous aluminas with amorphous XRD patterns have been also reported in previous studies.38,39 MA3 shows weak amorphous diffraction peaks accompanied with five crystalline peaks at 2θ of 18.89, 20.42, 27.95, 40.78 and 53.41°, which were identified as bayerite (PDF no. 770114). A crystalline bayerite pattern was also revealed from the Mo-target XRD (Fig. S1†).
 |
| | Fig. 3 Cu Kα XRD of patterns of the three mesoporous aluminas (°) indicates the diffraction peaks from bayerite. | |
3.2. Sorption studies
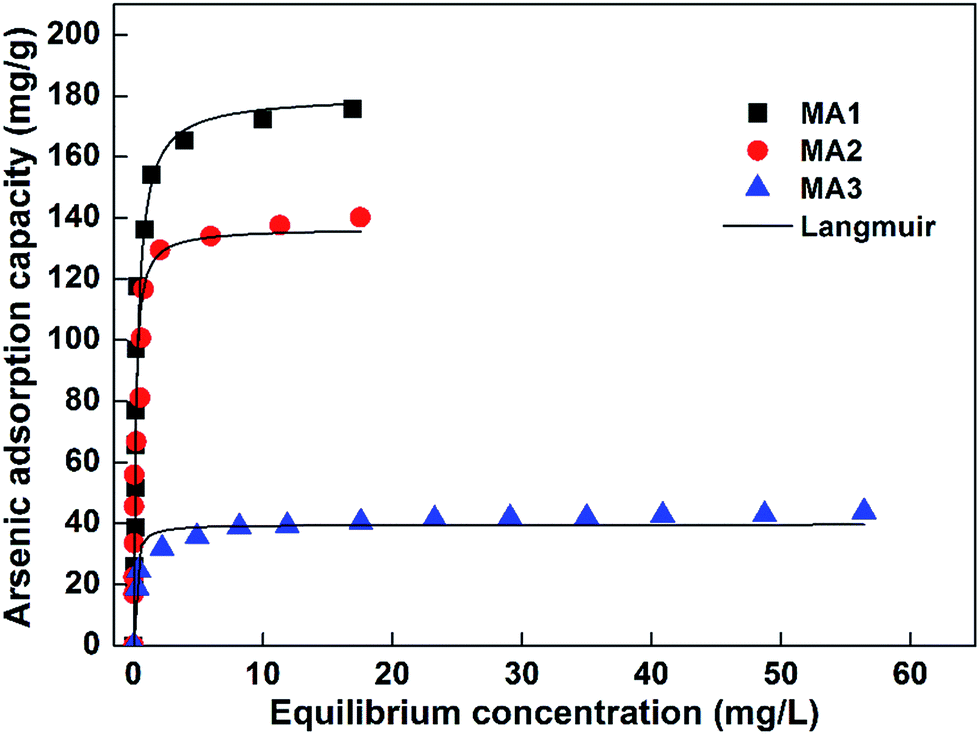
3.2.1. Adsorption isotherms. The adsorption isotherms of the three mesoporous aluminas are shown in Fig. 4. The As(V) uptake ability of MA1 is modestly higher than MA2, and is far larger than that of MA3. MA1 showed an inspiring maximum adsorption capacity of 175.7 mg g−1 at pH 5.0. This difference of MA1 versus MA2 and MA3 is more pronounced than indicated in Fig. 4, because the surface area of MA1 is less than that of MA2 and MA3. When the uptake ability is expressed by adsorption density, the amount of As(V) adsorbed per unit of surface area by MA1 is much higher. MA1 has a far smaller surface area of 166.7 m2 g−1 versus that of MA2 (543.4 m2 g−1) and a somewhat smaller surface area than MA3 (200.8 m2 g−1). The MA1 adsorption density is 1.41 × 10−20 mmol nm−2 versus 0.34 × 10−20 mmol nm−2 for MA2, and 0.29 × 10−20 mmol nm−2 for MA3, respectively. Both Langmuir and Freundlich models were applied to fit the isotherm data (Fig. 4 and Table S1†). The Langmuir fittings are better, giving higher correlation coefficients (R2) and lower root mean square of errors (RMSE) values than the Freundlich fittings, indicating that the Langmuir model may be more favorable compared to the Freundlich model based on statistical analysis. In addition, a performance comparison between the three MAs and various reported adsorbents, especially mesoporous aluminas, is provided in Table S2.† In all, MA1 was superior to many reported adsorbents.
 |
| | Fig. 4 Fitting the Langmuir model to the MA adsorption data. Initial As(V) concentrations, 6–70 mg L−1; adsorbent dose, 0.3 g L−1; total solution volumes, 100 mL; pH, 5.0 ± 0.1; temperature, 25 ± 1 °C, and shaking time, 48 h. | |
3.2.2. Adsorption kinetics. Kinetics of As(V) adsorption onto the three mesoporous aluminas at two different initial concentrations (∼18.0 mg L−1 and ~25.0 mg L−1, respectively) are shown in Fig. 5. The decay curves showed that As(V) adsorption on MA1 was quite fast at these concentrations. Most adsorption (>90%) by MA1 was achieved within 7 min versus 80 min for MA2. Adsorption on MA3 took longer (325 min) to reach equilibrium than on MA1 or MA2.
 |
| | Fig. 5 Fitting the MA adsorption data with the HSDM. Initial As(V) concentrations, ca. 18 and 25 mg L−1, respectively; adsorbent dose, 0.3 g L−1; total solution volumes, 1000 mL; pH, 5.0 ± 0.1; temperature, 25 ± 1 °C, and shaking time, 30 h. | |
3.2.3. Calculation of intraparticle diffusion coefficients. The concentration decay curves were fitted using the HSDM model. The results are presented in Fig. 5 and Table 2. The best fit curves are obtained by minimizing an objective function, the RMSE value between the experimental data and model estimations. The equation was given as,| |
 | (6) |
where CA,exp and CA,model are the experimental and model predictions of As(V) concentrations, respectively, obtained in the solution.
Table 2 Experimental conditions and mass transfer coefficients for As(V) adsorption on the MA adsorbents
| Adsorbents |
C0 (mg L−1) |
m (g) |
Ce (mg L−1) |
qe (mg g−1) |
Ds × 108 (cm2 s−1) |
RMSE |
| MA1 |
18.00 |
0.30 |
0.006 |
59.97 |
0.23 |
0.366 |
| 25.32 |
0.30 |
0.004 |
84.38 |
0.25 |
0.311 |
| MA2 |
18.07 |
0.30 |
0.010 |
60.20 |
0.22 |
0.723 |
| 25.40 |
0.30 |
1.176 |
80.74 |
0.23 |
0.603 |
| MA3 |
17.94 |
0.30 |
10.487 |
24.83 |
0.18 |
0.995 |
| 28.62 |
0.30 |
17.242 |
38.05 |
0.19 |
0.703 |
The low RMSE values indicated that the HSDM model fitted the concentration decay datasets reasonably well (Table 2). The mass transfer parameter in the HSDM model is Ds. Values of Ds ranged from 0.18 × 10−8 to 0.25 × 10−8 cm2 s−1. Ds decreased following an order of MA1 > MA2 > MA3 at the two initial As(V) concentrations. The Ds value increased for each mesoporous alumina at the higher initial As(V) concentration (∼25.0 mg L−1) versus the low concentration (∼18.0 mg L−1). The enlargement of Ds with higher As(V) concentrations results from the faster concentration gradient-driven intraparticle diffusion at the early stage of adsorption.
Fig. S2† presents the evolution of the concentration front profiles inside adsorbent particles at various times with initial As(V) concentrations at ∼25 mg L−1. Accordingly, Fig. 6 shows the post-processing of As(V) saturation (q/qe vs. t) within adsorbent particles at different times with the same initial As(V) concentrations. The dynamics of adsorption capacity depletion (or As(V) uptake ability utilization) was illustrated in a quite intuitive way. Solid phase concentration gradients within the three kinds of particles weakened gradually. It took 7 min to get 90% saturation of MA1 particles, 80 min for MA2 and 325 min for MA3, corresponding to the resulting concentration front profile evolutions inside the particles. It is quite obvious that MA1 has the fastest adsorption kinetics, followed by MA2, and the slowest was MA3.
 |
| | Fig. 6 Intraparticle two-dimensional profile of the concentration of the As(V) through the center of a single particle at various times at an initial As(V) concentration of ∼25 mg L−1; adsorbent dose, 0.3 g L−1; pH, 5.0 ± 0.1; shaking time, 30 h; temperature, 25 ± 1 °C. | |
In order to understand the kinetics of MA1 versus MA2 and MA3, a calculation of the effect of particle size on As(V) diffusion inside adsorbent particles was carried out. These results are presented in Fig. S3.† When the particle size was reduced (R, 3/4R, 1/2R and 1/4R) for each alumina, faster kinetics were achieved with progressively smaller particle sizes. In comparison to MA2 (134 μm) and MA3 (245 μm), MA1 has the minimum average particle size of 42 μm. This contributes to its fast As(V) mass transfer accompanied with the strong diffusion potential resulting from its high binding site density.
3.2.4. Effect of pH on adsorption. The effect of pH on As(V) adsorption by the three mesoporous adsorbents is shown in Fig. S4.† As(V) uptake on all three aluminas gradually increased as solution pH increased from 3.8 to 5.9, and then dropped between pH 5.9 and 9.8. MA1 is efficient over the investigated pH range with the least adsorption capacity loss when compared with MA2. MA1 was obviously superior to MA3. As(V) uptake by the three MAs versus pH (Fig. S4†) did not follow similar trends to the zeta potentials as they varied with solution pH (Fig. S5†). It suggested that the electrostatic force was not the dominant factor and another governing factor like chemical binding affinity also contribute to As(V) adsorption.
3.2.5. Treatment of simulated well water. Arsenic-contaminated well water usually contains multiple heavy metals and complex anions and cations. As(V) removal from simulated well water using the three adsorbents was evaluated and presented in Fig. S6.† The properties of the simulated well water are summarized in Table S3.† The residual arsenic could be lowered well below both the WHO guideline and Chinese regulation levels of 10 μg L−1 for As(V) in drinking water within 20 min, 65 min and 60 min for MA1, MA2 and MA3 with dosages of 0.1 g L−1, 0.1 g L−1 and 0.5 g L−1, respectively. These results clearly demonstrate that MA1 was the most effective of the three for As(V) removal when treating simulated well water. MA1 was regenerated by 0.01 M NaOH followed by 0.01 M HCl neutralization and reused at least for 5 cycles. At the 5th cycles, the capacity loss is 9.23% (Fig. S7†). In future practical application, powdered MA1 can either be granulated and be packed in a fixed bed, or be magnetically amended with magnetite and separated and reused in continuous stirred-tank reactors (CSTRs) with an external magnetic field installed. In all, MA1 is promising for arsenic mitigation and remediation in real applications.
3.3. The key properties accounting for As(V) sorption
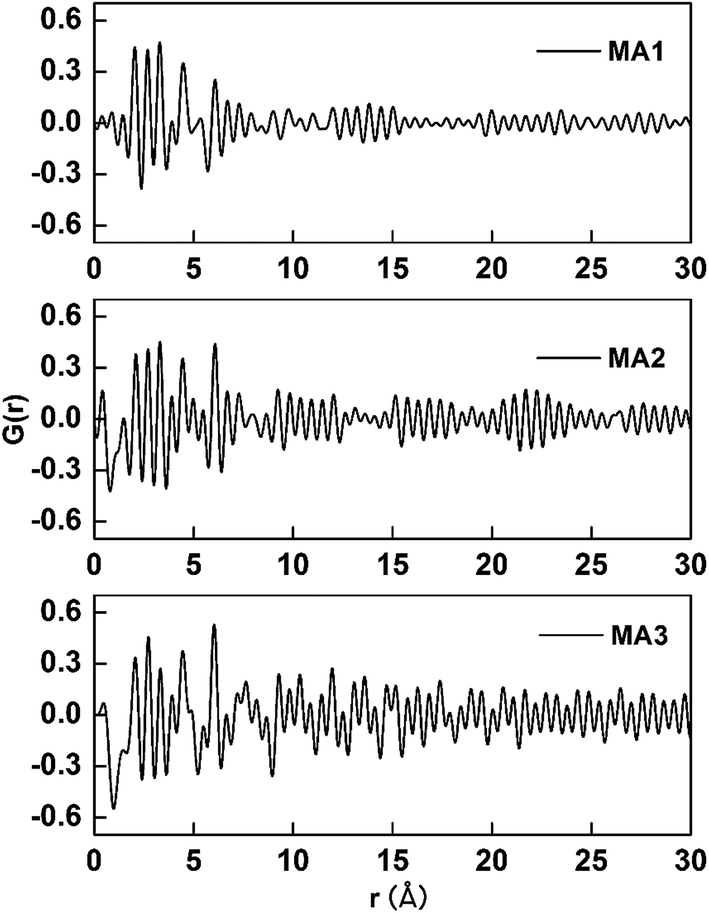
3.3.1. The effect of local atomic arrangements on As(V) adsorption. The PDF patterns of the three adsorbents are displayed in Fig. 7. The intensity of the peaks is explained by the multiplicity of the atom-pairs which included the number of atom-pairs at that particular distance and is weighted by the scattering power of the atoms in the pair. Peak broadening depends on the distance distribution around the average bond-length value, which results from thermal vibrations or disorder.40 Fig. 7 shows the amplitudes of the PDF signals attenuate with increasing r (Å) for all the three samples. The first peaks of the G(r) function correspond to the nearest neighbor atom pairs, the next nearest neighbor atom pairs, and next further pair distance. The value of the y-axis corresponds to the probability of finding two atoms separated by the x-axis distance, r (Å). The experimental PDF for MA3 is rich in well-defined structural features extending to the highest real-space distances (30 Å). This should be seen with a material possessing long-range atomic order and MA3 has the highest degree of crystallinity among the three. The PDFs for MA2 and MA1 are also rich in well-defined features but those features damp and vanish by approximately 13 and 8 Å, respectively. The order of long-range atomic arrangement obviously decreases in the order MA3 > MA2 > MA1, which is inversely correlated with the adsorption ability order of the three aluminas, where MA1 > MA2 > MA3. This means that strongly disordered atomic arrangements enhance the As(V) uptake abilities of these MAs.
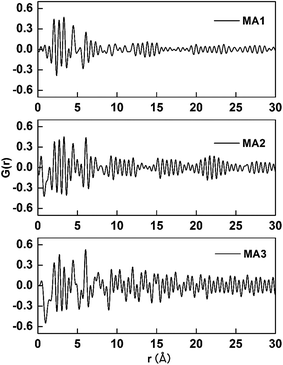 |
| | Fig. 7 The G(r) function deduced from the Mo-target X-ray source for the three MAs adsorbents. | |
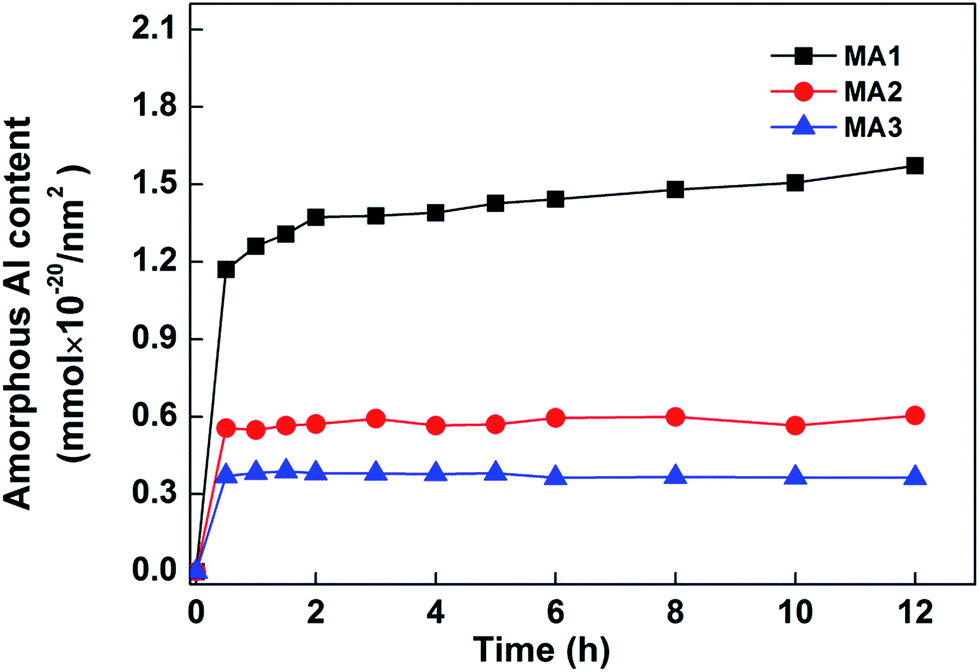
3.3.2. The effect of the aluminas' amorphous content fractions on As(V) adsorption. PDF results discussed earlier pointed out that the extent of structure disorder exhibited by MA samples affects their adsorption performance. Physical methods are not suitable to analyze quantitatively the amorphous content in aluminas. Instead a selective chemical extraction method was used to indirectly measure the amorphous content fractions and their effect on As(V) adsorption.41 This method indirectly determines the amount of the amorphous content present by measuring the complexation-promoted dissolution rate of metal oxides in the presence of ligands.42 For decades this method has been widely used to characterize the contents of amorphous Fe, Al, Mn and Si in soil and sediment.43,44 Amorphous complexes expose more reactive interfaces for dissolution than crystalline fractions. Amorphous regions originate from the microheterogeneities and irregularities at a microscopic scale in samples. These irregularities may be point defects, dislocations, microfractures, kinks, domain boundaries, corners, ledges and edges. In brief, the oxides, which have larger amorphous contents, will show faster dissolution rates than oxides with smaller amorphous contents.42 The complexation-promoted dissolution rate of the three MA samples using oxalic acid and its diammonium salt are presented in Fig. 8. MA1 has the fastest dissolution rate, followed by MA2 then MA3. MA1 has a far higher content of amorphous regions, while MA2 and MA3 have significantly smaller fractions. The complexation-promoted dissolution rates follow the same order as the adsorption capacities of the three MAs. Scale factors of 0.688 × 1020 g nm−2, 0.301 × 1020 g nm−2 and 0.633 × 1020 g nm−2 were calculated for the ratios of dissolution rates to the adsorption capacities for MA1, MA2 and MA3, respectively. In short, these results indicated the amorphous contents of MA samples are positively correlated with their adsorption capacities. Meanwhile, a particle size-dependent dissolution phenomenon has also been reported.45,46 This possibility could not be definitively excluded from the present result, so more research on this effect and on its role in the complexation promoted extraction results need to be carried out.
 |
| | Fig. 8 Al dissolution profiles of the three MA adsorbents in a solution containing (COOH)2·2H2O and (COONH4)2·H2O. The concentrations of (COOH)2·2H2O and (COONH4)2·H2O, were each 0.001 mol L−1; adsorbent dose, 0.5 g L−1; total solution volumes, 1000 mL; temperature, 50 ± 1 °C, and shaking time, 24 h; in the dark. | |
3.3.3. The effect of surface hydroxyl sites on As(V) adsorption. Metal hydroxyl groups on the surface of metal oxides are the most abundant and reactive adsorption sites responsible for anion adsorption.47 The amounts of surface hydroxyl sites on the three MA samples were determined using a titration method (see S2 in the ESI†). MA1 has the highest number of surface hydroxyl groups per unit weight (1.834 mmol g−1), followed by MA2 (0.356 mmol g−1), and then MA3 (0.064 mmol g−1). Clearly, a large quantity of titratable hydroxyl groups exist on MA1 despite its significantly lower surface area versus MA2 (Table 1). Using the BET surface areas of the three samples, the amounts of titratable surface hydroxyl sites were converted to the titratable surface hydroxyls per unit surface area (e.g. site densities) with a unit of sites per nm2. MA1 has the largest density of titratable surface sites (6.62 sites per nm2) versus MA2 (0.39 sites per nm2) and MA3 (0.19 sites per nm2). This order is consistent with the adsorption density order of the three mesoporous aluminas: MA1 (1.41 × 10−20 mmol As per nm2) versus MA2 (0.34 × 10−20 mmol As per nm2) and MA3 (0.29 × 10−20 mmol As per nm2). So the larger density of titratable surface hydroxyl sites correlates with the greater adsorption. In short, it is apparent that the MA samples' adsorption capacities correlate directly with their corresponding surface sites densities, and the difference of surface sites accounts for the performance differences. This makes good chemical sense since the surface hydroxyl groups' oxygen atoms could become bound to As(V) during adsorption. This was confirmed by XPS observations, since there were −0.29 eV, −0.67 eV and −0.10 eV shifts of the Al 2p binding energy peaks for As(V)-adsorbed MA1, MA2 and MA3 samples, respectively (see Fig. S8 and S4 in the ESI†).
3.3.4. The effect of Al–O coordination environment on As(V) adsorption. The 27Al solid state NMR of the three mesoporous aluminas are presented in Fig. 9. Three resonances from MA1 were observed around 8.2 ppm, 36.1 ppm and 68.4 ppm. These are due to octahedral-coordinated AlO6, pentahedral-coordinated AlO5 and tetrahedral-coordinated AlO4, respectively.48 One side band at 85.5 ppm also appeared. MA2 has two resonances at 6.4 ppm and 65.4 ppm and two sidebands at 104.1 ppm and −93.1 ppm. MA3 presents one resonance at 7.9 ppm and two sidebands at 105.9 ppm and −85.9 ppm. These spectra indicate that AlO6 is the only component observed for MA3, while MA2 has an additional small portion of AlO4 and MA1 contains all the three components (AlO4, AlO5 and AlO6). 27Al solid-state NMR clearly evidenced substantial Al–O coordination differences among the three mesoporous aluminas. AlO4 in γ-Al2O3 was reported to contain a substantial fraction of tetrahedrally coordinated Al atoms, and its concentration seems to correlate with its surface properties.40 Moreover, AlO4 has stronger Lewis acidity than AlO6. The existence of AlO5 motifs has not been reported for any of the crystalline Al2O3 polymorphs. An Al coordination number of five (AlO5) has been detected in amorphous and liquid alumina and the two have similar local structures.49 Five-coordinate aluminum is also considered as a Lewis acid center present in amorphous domains.50 Thus, the observation of AlO5 indicated the amorphous nature of the local structure of alumina and the presence of a Lewis acid center in the amorphous domain in the present study. Additional AlO5 plus AlO4 confer upon MA1 the strongest surface Lewis acidity among the three.51 This may contribute to and enhance As(V) adsorption on its surface.
 |
| | Fig. 9 27Al NMR spectra of the three mesoporous aluminas. Parts of the spectral region are magnified by a factor of 5, where the signals from AlO6, AlO5, AlO4 and sidebands are located around 7.3, 35.5, 68.1 and 81.5 ppm, respectively. | |
3.4. Property-performance summary of the MA samples
No consistent association or connection existed between the As(V) uptake ability of the three MAs with either BET surface area or microporosity. This key observation is understandable because the interface science involved in adsorption is quite complex, and many other surface and bulk properties might affect the adsorbent performances. In the present study, the following four features could account for the excellent As(V) adsorption ability of MA1 versus MA2 and MA3: (1) strongly disordered local atomic arrangement, (2) the existence of both tetrahedral AlO4 and pentahedral AlO5, (3) high surface adsorption site density and (4) large amorphous contents.
4. Conclusions
Three mesoporous aluminas were synthesized by facile chemical methods and used for As(V) removal from water. Performances in batch tests were evaluated. MA1 had the largest As(V) adsorption capacity of 175.7 mg g−1, which is superior to the other two MAs and many other reported adsorbents. MA1 shows the fastest adsorption kinetics, and its concentration decay curve was fitted using the HSDM model. The surface diffusion coefficient, Ds, for MA1 was determined to be 0.23 × 10−8 and 0.25 × 10−8 cm2 s−1 for initial As(V) concentrations of ∼18 mg L−1 and ∼25 mg L−1, respectively. Lowering solution pH enhanced As(V) adsorption on MAs. The outstanding performance of MA1 was explained by its unique features including a disordered state of local atomic arrangement from pair distribution function analyses, a large fraction of amorphous content determined by selective chemical extraction, abundant surface hydroxyl sites defined from surface titration, and the existence of AlO4 and AlO5 species demonstrated from 27Al solid-state NMR spectra. HSDM calculations indicated fast MA1 adsorption kinetics resulted from its small particle size as well as the strong diffusion potential resulting from its high As(V) binding site density.
Nomenclature
| C | Concentration of As(V) at t time (mg L−1) |
| C0 | Initial concentration of As(V) in aqueous solution (mg L−1) |
| Ce | Concentration of As(V) at equilibrium (mg L−1) |
| Cexp | Concentration of As(V) recorded in the experiments (mg L−1) |
| Cpred | Concentration of As(V) predicted with the simulation (mg L−1) |
| Ds | Intraparticle surface diffusion coefficient (cm2 s−1) |
![[q with combining macron]](https://www.rsc.org/images/entities/i_char_0071_0304.gif) | Average mass of As(V) adsorbed per unit mass of adsorbent (mg g−1) |
| q | Mass of As(V) adsorbed per unit mass of adsorbent (mg g−1) |
| qe | Mass of As(V) adsorbed per unit mass of adsorbent at equilibrium (mg g−1) |
| m | Mass of adsorbent (g) |
| V | Volume of the As(V) solution (L) |
| Rp | Radius of MA particle (cm) |
| r | Distance in radial direction of MA particle (cm) |
| T | Time (min) |
Greek letters
| ρp | Apparent particle density (g mL−1) |
| εp | Void fraction of mesoporous alumina |
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (2015ZCQ-HJ-02) and the Fundamental Research Funds for the Central Universities (TD-JC-2013-3).
References
- P. L. Smedley and D. G. Kinniburgh, United Nations synthesis report on arsenic in drinking water, World Health Organization, Geneva, Switzerland, 2001, pp. 1–61 Search PubMed.
- S. C. Wilson, P. V. Lockwood, P. M. Ashley and M. Tighe, Environ. Pollut., 2010, 158, 1169–1181 CrossRef CAS PubMed.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- V. M. Boddu, K. Abburi, J. L. Talbott, E. D. Smith and R. Haasch, Water Res., 2008, 42, 633–642 CrossRef CAS PubMed.
- H. K. Hansen, P. Nunez and R. Grandon, Miner. Eng., 2006, 19, 521–524 CrossRef CAS.
- M. Borho and P. Wilderer, Water Sci. Technol., 1996, 34, 25–31 CrossRef CAS.
- L. Dambies, Sep. Sci. Technol., 2004, 39, 603–627 CrossRef CAS.
- A. U. Baes, T. Okuda, W. Nishijima, E. Shoto and M. Okada, Water Sci. Technol., 1997, 35, 89–95 CrossRef CAS.
- T. S. Y. Choong, T. G. Chuah, Y. Robiah, F. L. Gregory Koay and I. Azni, Desalination, 2007, 217, 139–166 CrossRef CAS.
- J. Hong, Z. Zhu, H. Lu and Y. Qiu, Chem. Eng. J., 2014, 252, 267–274 CrossRef CAS.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- A. S. Chen, T. J. Sorg and L. Wang, Water Res., 2015, 77, 85–97 CrossRef CAS PubMed.
- Y. Jeong, M. Fan, S. Singh, C. Chuang, B. Saha and J. Hans van Leeuwen, Chemical Engineering and Processing: Process Intensification, 2007, 46, 1030–1039 CrossRef CAS.
- J. Shen, Z. Li, Y.-N. Wu, B. Zhang and F. Li, Chem. Eng. J., 2015, 264, 48–55 CrossRef CAS.
- C. Han, H. Pu, H. Li, L. Deng, S. Huang, S. He and Y. Luo, J. Hazard. Mater., 2013, 254–255, 301–309 CrossRef CAS PubMed.
- S. Jagtap, M. K. N. Yenkie, N. Labhsetwar and S. Rayalu, Microporous Mesoporous Mater., 2011, 142, 454–463 CrossRef CAS.
- M. J. Yu, X. Li and W. S. Ahn, Microporous Mesoporous Mater., 2008, 113, 197–203 CrossRef CAS.
- W. Cai, L. Tan, J. Yu, M. Jaroniec, X. Liu, B. Cheng and F. Verpoort, Chem. Eng. J., 2014, 239, 207–215 CrossRef CAS.
- Y. B. Sun, C. L. Chen, X. L. Tan, D. D. Shao, J. X. Li, G. X. Zhao, S. B. Yang, Q. Wang and X. K. Wang, Dalton Trans., 2012, 41, 13388–13394 RSC.
- S. Ghosh and M. K. Naskar, J. Am. Ceram. Soc., 2014, 97, 100–106 CrossRef CAS.
- A. K. Patra, A. Dutta and A. Bhaumik, J. Hazard. Mater., 2012, 201–202, 170–177 CrossRef CAS PubMed.
- J. Hao, M. Han, C. Wang and X. Meng, Microporous Mesoporous Mater., 2009, 124, 1–7 CrossRef CAS.
- P. Pillewan, S. Mukherjee, T. Roychowdhury, S. Das, A. Bansiwal and S. Rayalu, J. Hazard. Mater., 2011, 186, 367–375 CrossRef CAS PubMed.
- S. M. Maliyekkal, L. Philip and T. Pradeep, Chem. Eng. J., 2009, 153, 101–107 CrossRef CAS.
- W. Li, C. Y. Cao, L. Y. Wu, M. F. Ge and W. G. Song, J. Hazard. Mater., 2011, 198, 143–150 CrossRef CAS PubMed.
- B. Chen, Z. Zhu, Y. Guo, Y. Qiu and J. Zhao, J. Colloid Interface Sci., 2013, 398, 142–151 CrossRef CAS PubMed.
- M. Jang, J. K. Park and E. W. Shin, Microporous Mesoporous Mater., 2004, 75, 159–168 CrossRef CAS.
- S. J. Billinge and M. Kanatzidis, Chem. Commun., 2004, 749–760 RSC.
- F. Atassi, C. Mao, A. S. Masadeh and S. R. Byrn, J. Pharm. Sci., 2010, 99, 3684–3697 CrossRef CAS PubMed.
- P. Juhas, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef CAS.
- K. H. Chu, J. Water Process Eng., 2014, 3, 117–122 CrossRef.
- G. MacKay, Use of adsorbents for the removal of pollutants from wastewaters, CRC, Boca Raton, 1996 Search PubMed.
- G. A. Heeter and A. I. Liapis, J. Chromatogr. A, 1997, 760, 55–69 CrossRef CAS.
- Q. Zhang, J. Crittenden, K. Hristovski, D. Hand and P. Westerhoff, Water Res., 2009, 43, 1859–1866 CrossRef CAS PubMed.
- Y. L. Yeh, C. M. Liao, J. S. Chen and J. W. Chen, Appl. Math. Model., 2001, 25, 593–611 CrossRef.
- A. Schnepf, T. Schrefl and W. W. Wenzel, J. Plant Nutr. Soil Sci., 2002, 165, 713–718 CrossRef CAS.
- J. Shi, Z. Tang, Z. Jin, Q. Chi, B. He and G. Jiang, Anal. Chim. Acta, 2003, 477, 139–147 CrossRef CAS.
- R. Zhao, F. Guo, Y. Hu and H. Zhao, Microporous Mesoporous Mater., 2006, 93, 212–216 CrossRef CAS.
- Z.-X. Sun, T.-T. Zheng, Q.-B. Bo, M. Du and W. Forsling, J. Colloid Interface Sci., 2008, 319, 247–251 CrossRef CAS PubMed.
- L. Samain, A. Jaworski, M. Edén, D. M. Ladd, D. Seo, F. Javier GarciaGarcia and U. Häussermann, J. Solid State Chem., 2014, 217, 1–8 CrossRef CAS.
- T. Chao and L. Zhou, Soil Sci. Soc. Am. J., 1983, 47, 225–232 CrossRef CAS.
- G. T. Burstein, Corros. Sci., 1997, 39, 1499–1500 CrossRef CAS.
- C. Gleyzes, S. Tellier and M. Astruc, TrAC, Trends Anal. Chem., 2002, 21, 451–467 CrossRef CAS.
- A. Tessier, P. G. Campbell and M. Bisson, Anal. Chem., 1979, 51, 844–851 CrossRef CAS.
- W. Vogelsberger, J. Schmidt and F. Roelofs, Colloids Surf., A, 2008, 324, 51–57 CrossRef CAS.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- H. Tamura, A. Tanaka, K. Mita and R. Furuichi, J. Colloid Interface Sci., 1999, 209, 225–231 CrossRef CAS PubMed.
- N. Kim, R. Bassiri, M. M. Fejer and J. F. Stebbins, J. Non-Cryst. Solids, 2014, 405, 1–6 CrossRef CAS.
- A. H. Tavakoli, P. S. Maram, S. J. Widgeon, J. Rufner, K. van Benthem, S. Ushakov, S. Sen and A. Navrotsky, J. Phys. Chem. C, 2013, 117, 17123–17130 CAS.
- L. Xue, J. Wei, D. Zhang, X. Li and X. Liu, J. Porous Mater., 2015, 22, 1119–1126 CrossRef CAS.
- J. S. Valente, E. López-Salinas, X. Bokhimi, J. Flores, A. M. Maubert and E. Lima, J. Phys. Chem. C, 2009, 113, 16476–16484 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14408j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Charles U. Pittman Jr.c and
Meng Hud
b,
Charles U. Pittman Jr.c and
Meng Hud