
Photoresponsive nanostructured membranes†
P. Madhavana,
B. Sutisnab,
R. Sougratc and
S. P. Nunes*a
aKing Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering Division (BESE), 23955-6900 Thuwal, Saudi Arabia. E-mail: suzana.nunes@kaust.edu.sa
bKing Abdullah University of Science and Technology (KAUST), Physical Science and Engineering Division (PSE), 23955-6900 Thuwal, Saudi Arabia
cKing Abdullah University of Science and Technology (KAUST), Advanced Nanofabrication Imaging and Characterization Core Lab, 23955-6900 Thuwal, Saudi Arabia
First published on 26th July 2016
Abstract
The perspective of adding stimuli-response to isoporous membranes stimulates the development of separation devices with pores, which would open or close under control of environment chemical composition, temperature or exposure to light. Changes in pH and temperature have been previously investigated. In this work, we demonstrate for the first time the preparation of photoresponsive isoporous membranes, applying self-assembly non-solvent induced phase separation to a new light responsive block copolymer. First, we optimized the membrane formation by using poly(styrene-b-anthracene methyl methacrylate-b-methylmethacrylate) (PS-b-PAnMMA-b-PMMA) copolymer, identifying the most suitable solvent, copolymer block length, and other parameters. The obtained final triblock copolymer membrane morphologies were characterized using atomic force and electron microscopy. The microscopic analysis reveals that the PS-b-PAnMMA-b-PMMA copolymer can form both lamellar and ordered hexagonal nanoporous structures on the membrane top layer in appropriate solvent compositions. The nanostructured membrane emits fluorescence due to the presence of the anthracene mid-block. On irradiation of light the PS-b-PAnMMA-b-PMMA copolymer membranes has an additional stimuli response. The anthracene group undergoes conformational changes by forming [4 + 4] cycloadducts and this alters the membrane's water flux and solute retention.
Introduction
Stimuli-responsive materials change their physicochemical properties under the influence of different environmental conditions, such as pH, temperature, light, electric and magnetic fields.1–5 The main factor guiding separation in porous membranes is size difference. Transport in biological systems is in part controlled by a pore gate response to pH, ionic strength or biological recognition. This is the basis for the development of biomedical devices aiming at controlled insulin delivery. Actuators as artificial muscles respond to electric impulses. The development of stimuli-responsive membranes with tunable properties provides additional opportunities in flux modulations, and separation selectivity control for various advanced applications, e.g. controlled drug release and sensors for biomedical applications.6–11 If stimuli responsive functional groups are part of the membrane material,12–16 conformational changes might occur, leading to changes in the degree of swelling/deswellling, with consequences on the membrane permeability and selectivity. Temperature and pH response have been more frequently investigated.1–3 We propose here a strategy to develop photoresponsive membranes with tunable properties and potential applications as optical gates for controlled drug delivery, diagnostics or water-based separation triggered by light.10Highly ordered membranes were prepared by combining self-assembly of block copolymer and non-solvent induced phase separation. Membranes with high water flux and good solute rejection have been previously prepared by this method, with excellent pH responsive behavior.17–21 Most of the development was done using pyridine or polyacrylic acid blocks. Nanopores of PS-b-P4VP membrane close and open when subject to external pH conditions.17 The nitrogen atom in the basic pyridine group protonates under acidic condition and leads to pore closing. Pyridine is deprotonated under basic conditions, leading then to reversibly opened pores. On the other hand PS-b-PAA copolymer porous structures have close pores in basic pH and open at low pH. Our effort is targeted towards exploring response to light instead of pH, extending the possibilities of applications, keeping high order and porosity. This is the first time that light response is combined to block copolymer membranes prepared by non-solvent induced phase separation. To achieve the light responsive behaviour, we chose poly(styrene-b-anthracene methyl methacrylate-b-methylmethacrylate) (PS-b-PAnMMA-b-PMMA) triblock copolymer as membrane forming material. The amphiphilic character of the block copolymer led to an ordered self-assembled morphology and the anthracene methyl methacrylate block provided the light response. Anthracene and its derivatives have been widely used as organic optoelectronic materials due to their high fluorescence quantum yields.22–24 Anthracene moiety can be transformed into photodimers upon long-wave UV irradiation (>350 nm) to give [4 + 4] cycloadducts and that the photodimer can be restored to original monomers upon exposure to shortwave UV light (254 nm). This photochromism has been studied extensively for many intermolecular and intramolecular processes.25–27 For example, Zheng and co-workers28 developed photoreversible poly(ethylene glycol)-anthracene based hydrogel through the photodimerization of anthracene groups. Rameshbabu et al. prepared photo patternable anthracene, covalently connected through a methylene bridge, which showed significantly higher fluorescence compared to monomeric anthracene.29 Hargreaves et al., reported the synthesis of poly(9-anthrylmethyl methacrylate) polymers, which have a strong tendency to be photoreactive. The use of anthracene moieties as cross-linking sites to produce photoreversible organogels has been also reported.30 To alter the membrane properties with response to light, a key strategy is to utilize the photochromism behaviour of anthracene group as one of the blocks in the bulk block copolymer membrane. In the present study, we report the formation of lamella and ordered hexagonal nanopores, by using casting solutions in selective solvents, using PS-b-PAnMMA-b-PMMA triblocks. We demonstrate the membrane light responsive behaviour.
Experimental
Materials
Poly(styrene-b-anthracene methylmethacrylate-b-methylmethacrylate) (PS-b-PAnMMA-b-PMMA) triblock copolymer with two different block length P4206 (44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-b-1200-b-45000), P4205 (43000-b-1500-b-105000) and poly(styrene-b-methylmethacrylate) (PS-b-PMMA) diblock copolymer P5543-SMMA (160000-b-160000) were purchased from Polymer Source, Inc., Canada. Dimethyl formamide (DMF), acetonitrile, dimethylacetamide (DMAc), acetone, tetrahydrofuran (THF) and bovine serum albumine were purchased from Sigma Aldrich.
000-b-1200-b-45000), P4205 (43000-b-1500-b-105000) and poly(styrene-b-methylmethacrylate) (PS-b-PMMA) diblock copolymer P5543-SMMA (160000-b-160000) were purchased from Polymer Source, Inc., Canada. Dimethyl formamide (DMF), acetonitrile, dimethylacetamide (DMAc), acetone, tetrahydrofuran (THF) and bovine serum albumine were purchased from Sigma Aldrich.
Membrane preparation
Nanostructured membranes were mostly cast from polymer solutions containing 18 wt% PS-b-PAnMMA-b-PMMA block copolymer (except in Fig. S2†). The block copolymers with different solvent systems were stirred at room temperature for 24 h, before forming the nanostructured membranes. Then the block copolymer solutions were cast on a glass plate using a casting knife with 200 μm air gap. The solvent was allowed to evaporate for different time intervals and the films were immersed in de-ionized water at room temperature.Field emission scanning electron microscopy (FESEM)
The surfaces of the membranes were imaged using FEI Quanta 600 Field Emission Scanning Electron Microscope. Imaging was carried out at 5 kV with a working distance of 10 mm. Then the membrane samples were sputter-coated with iridium (3 nm) under argon atmosphere and the membrane samples were mounted on aluminium stubs, using aluminium tape.Atomic force microscopy (AFM)
AFM analysis was performed using an Agilent microscope (Model 5400) in tapping mode. The tip was used with spring constant 3 N m−1, resonant frequency 76–263 kHz. Block copolymer membranes were imaged after drying.Transmission electron microscopy (TEM)
The membrane cross section was examined using TEM. Images were obtained on a Tecnai 12 (FEI) operating at 120 keV. The membranes were embedded in a low-viscosity epoxy resin (Agar R1165) and cured at 60 °C for 24 h. Ultra thin sections (80 nm) were prepared with an ultramicrotome (Leica EM UC6) and placed on a carbon-coated copper grid. Membrane samples were exposed to RuO4 vapors, which preferentially stain the aromatic blocks of the membrane before imaging. RuO4 was prepared by the following procedure.31 Sodium periodate (1.28 g) was dissolved in 100 mL of deionized water (25 °C) and cooled to 1 °C. Hydrated ruthenium dioxide (0.6 g) was then added to the chilled aqueous solution of NaIO4. As RuO2·xH2O began to dissolve, golden yellow RuO4 was formed.Water flux and retention measurement
The membranes were kept in water after manufacture. Pure water flux was measured using an Amicon dead end cell at 1 bar N2 pressure. Membranes were cut into circular samples of about 5 cm.2 Before irradiation the membrane water flux was measured and then the wet membrane was kept in water bath and irradiated in the UV light (8 watt at a distance of 72 mm) at 365 nm for 24 h. After irradiation the membrane water flux was measured again. Then the irradiation procedure was repeated at 264 nm for 6 h and the water flux was again determined. The permeance was calculated by using the following eqn (1)
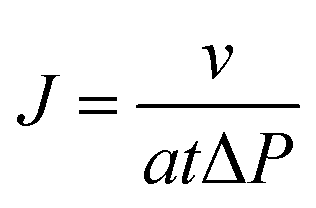 | (1) |
 | (2) |
Fluorescence measurement
Fluorescence micrographs of the nanostructured film were captured using epifluorescence optical microscope (Olympus B X61) with a filter at 495/520 nm. Photodimerization was carried out for the block copolymer membranes obtained by phase inversion method. The membranes were kept in water and irradiated upon UV lamp at λ = 365 nm for 24 h and then dried the membranes for fluorescence measurements. Fluorescence measurements for the solid film were carried out using a Cary Eclipse fluorescence spectrophotometer (FL0910M008) at the ambient temperature. The same procedure was repeated for the membranes irradiated at λ = 264 nm. For all measurements, the films were excited at λex = 370 nm and their corresponding emission wavelength was monitored from λex = 370 nm. Both excitation and emission slit width were 2.5 nm for all measurement.Results and discussion
The first step in the membrane development is the achievement of a regular morphology with pores in the size scale adequate for the targeted separation. The morphology is very sensitive to the solvent quality as demonstrated by Fig. 1. The key steps for the preparation of nanostructured block copolymer membranes by phase inversion are: (i) the self-assembly of block copolymers in solution (ii) partial evaporation after casting the block copolymer solution on a substrate and (iii) immersion in a non-solvent (water in this case). The manufacture of highly ordered nanoporous membranes has been previously demonstrated by using the following block copolymers: PS-b-P4VP,19 PS-b-PEO,32 and PI-b-PS-b-P4VP.20 The pore formation in block copolymer membranes has been recently reviewed33 and discussed in detail. Here for the first time PS-b-PAnMMA-b-PMMA triblock copolymer was used for membrane preparation. The anthracene block gives unique properties as light responsive membrane. | ||
| Fig. 1 FESEM images of membranes surfaces cast from 18 wt% PS44k-b-PAnMMA1.2k-b-PMMA45k copolymer solutions in (a) DMAc, THF or acetone; (b) 1/1 DMAc/acetone, DMAc/THF or THF/acetone; (c) 35/29/18 wt% DMAc/acetone/THF with 10 s evaporation time; (d) 35/29/18 wt% DMAc/acetonitrile/THF with 20 s evaporation time. | ||
We recognized the importance of the micelle formation in solution to obtain a membrane with ordered isopores in hexagonal arrangement.18 As shown in Fig. 2, solvents able to form well-defined micelles in solution will give rise to regular pores, while swollen deformable micelles do not lead to order. To attain a desirable morphology, the block copolymer self-assembly into micelles must be tuned by choosing appropriate solvents suitable for selective interaction with the copolymer blocks. The membrane casting solution contained 18 wt% of PS-b-PAnMMA-b-PMMA copolymer in different solvent mixtures and immersed in water. Solvent-non-solvent exchange immediately takes place, leading to structure immobilization and pore formation. The membrane morphology was analysed by different methods of electron and atomic force microscopy. Fig. 1 compares field emission scanning electron microscopy (FESEM) images of PS44k-b-PAnMMA1.2k-b-PMMA45k triblock copolymer membranes, obtained with the same copolymer concentration (18 wt%) and different solvents or solvent mixtures. The solvents were chosen based on the Hansen solubility parameters given in Table S1.† A first requirement is the solvent miscibility with water to enable an effective solvent exchange and consequent phase separation to consolidate the pore formation. Fig. 1a shows the FESEM images of triblock copolymer membranes prepared using single solvents: DMAc, THF and acetone. Their total solubility parameters (δ) values are close to those of PS and PMMA blocks. But by analysing the individual contributions, particularly relative to polarity and hydrogen bonds, all solvents are slightly better for the PMMA block than for PS. The FESEM images for membranes prepared using single solvents did not show any ordered morphology. Single solvents do not lead in this case to a pronounced ordered self-assembly in solution. THF solubility parameters do not clearly favour one of the blocks and might not even stimulate micelle formation, leading to a surface without visible morphological features. Acetone is the one with the most distinguished interaction with each block, being more favourable for PMMA, but no order was observed either. Fig. 2 shows transmission electron microscopy (TEM) images of micelles formed in diluted solutions in each of the single solvents. It is clearly seen that THF does not favour the formation of regular micelles. Only swollen aggregates could be observed. If micelles are formed in a solvent, which highly swells the corona, its deformation disfavours the achievement of membranes with ordered porous structure. Acetone is the solvent with best-structured micelles, which are interconnected in arrays.
 | ||
| Fig. 2 TEM images of PS44k-b-PAnMMA1.2k-b-PMMA45k diluted solutions in DMAc, THF and acetone as single solvents and a mixture of them with 35/29/18 wt% ratio. | ||
Fig. 1b shows the morphology obtained, when binary solvents, DMAc/acetone, DMAc/THF or THF/acetone, are used. Some indication of lamellar morphology is seen for DMAc/THF. Globular structures are seen for DMAc/acetone, without preferential order. DMAc/acetone is probably the most selective combination, which might promote micelle formation with PS cores, but this is not enough to lead to regular surface morphology. When we used a ternary solvent mixture, the membrane surface was highly ordered with cylinders are formed. Micelles formed in dilute solutions in the ternary solvent are more compact than in the single solvents. They are spherical, well-formed and interconnected forming strings. The ternary solvent system was then cast and subjected to different evaporation times before immersion in water. Fig. 3 shows the results for evaporation times from 5 to 40 s. No order is seen with 5 s evaporation, whereas from 10 to 30 s very ordered lamellae or cylinders are formed. Longer evaporation leads to less order. The boiling points of acetone, THF and DMAc, increase from 56, 66 to 165 °C. Even short evaporation would lead to enrichment of DMAc in the cast solution, which is better for PMMA. In order to investigate the effect of block length we kept the ternary solvent system (35/29/18 wt% DMAc/acetone/THF), polymer concentration (18 wt%) and other optimized casting conditions, using now a copolymer with the double length of PMMA block: PS43k-b-PAnMMA1.5k-b-PMMA105k. Fig. 1c and d and 4c compare the morphologies of membranes cast from the two copolymers solutions under similar conditions using ternary solvent mixtures. While surface-aligned cylinders are clearly seen for the copolymer with shorter PMMA block in DMAc/acetone/THF (Fig. 1c), a different morphology is observed with the longer PMMA block in the same solvent mixture (Fig. 4c, left). We then replaced acetone by acetonitrile in the ternary solvent mixture for PS43k-b-PAnMMA1.5k-b-PMMA105k (higher PMMA content). Acetonitrile is a much more polar solvent. Spherical and very regular pores with hexagonal order were obtained with 20 s evaporation time, which was the best-optimized condition, as shown in Fig. 4c (right) and S1.† Single solvents and binary solvent mixtures were also investigated for PS43k-b-PAnMMA1.5k-b-PMMA105k. The copolymer was not soluble in acetonitrile alone; the morphologies obtained for single and binary systems are also shown in Fig. 4, confirming that the ternary solvent mixture is the most convenient one. Fig. 1d shows the morphology of a PS44k-b-PAnMMA1.2k-b-PMMA45k triblock copolymer membrane analogously cast from solution in DMAc/acetonitrile/THF with 20 s evaporation time. A highly porous structure was obtained but with low order than for the higher molecular weight copolymer in analogous conditions. We investigated the effect of polymer concentration on the selective ternary solvents (Fig. S2†). 18 wt% was the best-optimized concentration for both copolymer systems.
 | ||
| Fig. 3 FESEM images of membranes surfaces cast from 18 wt% PS44k-b-PAnMMA1.2k-b-PMMA45k copolymer solution in 35/29/18 wt% DMAc/acetone/THF with different evaporation times. | ||
 | ||
| Fig. 4 FESEM images of membranes surfaces cast from 18 wt% PS43k-b-PAnMMA1.5k-b-PMMA105k copolymer solutions in (a) DMAc or THF; (b) 1/1 DMAc/THF, DMAc/acetonitrile or THF/acetonitrile; (c, right) 35/29/18 DMAc/acetonitrile/THF with 20 s evaporation time and (c, left) DMAc/acetone/THF with 10 s evaporation time. | ||
Fig. 5 shows the surfaces of optimized membranes imaged by atomic force microscopy (AFM) and the cross-sections of the corresponding membranes imaged by TEM. This confirms the ordered structures previously observed by FESEM. The cross sections images for PS44k-b-PAnMMA1.2k-b-PMMA45k membranes prepared from solution in DMAc/acetone/THF show cylinders parallel to the surface on the top layer. At positions far from the surface a mixture of elongated structures and spherical assemblies is observed, indicating that cylinders (and not lamellae) are the predominant assembly geometry. For the PS43k-b-PAnMMA1.5k-b-PMMA105k membrane cast from solution in DMAc/acetonitrile/THF spherical micelles are seen not only close to the surface but overall in the membrane structure. RuO4 preferentially stains the blocks with aromatic rings, in this case PS and the short anthracene-containing blocks. By taking into account the solubility parameter values in Table S1,† PS should constitute the micelle cores. Changing the block length affects the entropic constrains of block assembly organization. Shorter PMMA blocks lead to cylindrical morphology, while longer PMMA blocks lead to spherical features. In a simplified illustration close-packed spherical micelles, as shown in Fig. 6, should prevail for PS44k-b-PAnMMA1.2k-b-PMMA105k, while cylindrical micelles are predominant in PS43k-b-PAnMMA1.5k-b-PMMA45k solutions. Periodic morphologies in block copolymers are strongly influenced by a competition between interface curvature and chain-packing conformation.34
 | ||
| Fig. 5 (a, b) Surface AFM images and (c, d) cross-section TEM images of (a, c) PS43k-b-PAnMMA1.5k-b-PMMA45k and (b, d) PS44k-b-PAnMMA1.2k-b-PMMA105k membranes: images of the top layers immediately close to the surface and at deeper locations. | ||
 | ||
| Fig. 6 Self-assembly of PS-b-PAnMMA-b-PMMA copolymers: (a) anthracene block dimerization under irradiation; (b, c) organization from solution bulk to membrane surface as (b) spherical and (c) cylindrical micelles. | ||
Solvent–block interaction in the solution bulk is important, but is not the only factor driving the morphology of the membrane top layer. Solution–air (or vapour) interfacial tension, the surface energy of each block and the block–block segregation strength are as relevant. When the solution is cast, a large surface area is exposed and the contribution of surface energy becomes stronger. A periodic morphology known as perforated lamellae,35–38 which is more common in thin films, could be related to the membrane formation with the high molecular weight copolymer. In copolymer systems with strong segregation this morphology is not favoured, but the right solvent–blocks interactions and surface energy could favour it, when the solution with ordered spherical micelles is cast. Immersion in water leads to abrupt solvent-non-solvent exchange with solidification and kinetic trapping of polymer structures, which might be metastable and in other conditions would vanish in favour of equilibrium morphologies. Fig. 5 shows cylinders parallel to the membranes surface for copolymers with shorter PMMA segments. When cylindrical assemblies are formed in the bulk, they often lay parallel to the surface, instead of orthogonal. Changes in cylinders orientation can be promoted by exposing the solution to different vapours, a procedure, which affects the interfacial energy at the air/film interface. If the surface energies of the constituting blocks are not strongly different the largest block might tend to accumulate on the surface and an entropic balance will guide the placement of the smaller block. This can not only change cylinders orientation, but also change the predominant morphology from cylinders to lamellae or cylinders to perforated lamellae.
Solvent annealing has been a successful tool to control morphology, particularly in thin films, as explored by different groups and revised by Hamley39 and Albert.40 When solid thin films are annealed, the solvent plays an important role, increasing the polymer mobility, kinetically enabling an energetically favourable transition. In our case we have morphology control in solution and mobility is already much higher than in a solid film. The solvent has the role of affecting the thermodynamically driven self-assembly in the bulk and the solution–air (vapour) interfacial energy. In this paper the copolymer containing anthracene was chosen because of the possibility of fluorescence emission. To demonstrate this behaviour, the membranes were imaged using a fluorescence microscope, after irradiation at 365 nm.
Fig. 7a shows the fluorescence micrographs of PS160k-b-PMMA160k (no anthracene moiety), PS44k-b-PAnMMA1.2k-b-PMMA45k, and PS43k-b-PAnMMA1.5k-b-PMMA105k membranes, prepared by phase inversion. The fluorescence micrographs of PS-b-PMMA did not show any blue emission because there is no fluorescent group. The other two images show homogeneous blue emission due to the fluorescence of anthracene present in the block copolymer. To investigate the reversibility of the dimerization, the dimerized membranes were then irradiated with light at 264 nm, a wavelength, which should not induce fluorescence. Fluorescence spectra of PS43k-b-PAnMMA1.5k-b-PMMA105k membranes with 365 nm and 264 nm irradiation are depicted in Fig. 7b and c. The reversible reaction (i.e., the conversion of cycloadduct to anthracene moiety) occurred after 2 h of irradiation at 264 nm. The conversion of cycloadduct to anthracene was maximal at 6 h of irradiation. Further irradiation at 264 nm led again to formation of cycloadduct. It was observed from Fig. 7, that the reversible reaction occurred after 5 or 6 h of irradiation and the conversion was only 50% achieved compared to the original non irradiated membrane, indicating that the anthracene moiety was only partially recovered. Anthracene is a polycyclic aromatic hydrocarbon, which undergoes photo-dimerization on exposure to light or heat and is also able to emit fluorescence if properly excited.29,30,41 Here in this work, the membranes were prepared by triblock copolymers containing anthracene moiety as one of the mid-block between PS and PMMA. The photochemical dimerization occurs upon long wavelength UV irradiation (>350 nm) to give [4 + 4] cycloadducts across the 9,10 positions of anthracene (Fig. 6). The dimerization reaction is reversible and the photo-dimers can be dissociated into the monomers, after heating or irradiation with other wavelength. Photo-dimerization occurs both in solid and liquid state. It is obviously faster in liquid state and is highly dependent on the relative position and orientation of the monomers.42 To trigger the dimerization, we carried out the photo-dimerization reaction upon irradiating with UV light at 365 nm. The block copolymer membranes were kept in water and directly exposed to UV lamp at 365 nm. Fig. 7b and c show the fluorescence spectra obtained for PS43k-b-PAnMMA1.5k-b-PMMA105k at various irradiation times and their reversible switching behaviour.
 | ||
| Fig. 7 (a) Fluorescence micrograph of PS-b-PMMA, PS44k-b-PAnMMA1.2k-b-PMMA45k and PS43k-b-PAnMMA1.5k-b-PMMA105k membranes; (b, c) fluorescence spectra of PS43k-b-PAnMMA1.5k-b-PMMA105k membranes with (b) 365 nm and (c) 264 nm irradiation. | ||
The distinct fluorescence bands for anthracene have peaks at about 385, 405, 420 and 445 nm. After irradiation during 0.5 h at 365 nm, the anthracene moiety converted into [4 + 4] cycloadducts and the fluorescence intensity decreased to half compared to that of the original membrane. This clearly shows that the photo-dimerization was fast even in the solid state. On further irradiation up to 24 h at 365 nm the intensity decrease even more. The dimerization process could be either intermolecular or intramolecular. The main requirement is that the anthracene groups should be in close proximity to react. The reaction is reversible, as shown in Fig. 7c. We anticipated that these changes could affect also permeability and selectivity. A flux decline indicates that conformational changes affected the pore size. When the dimerization reaction takes place between different chains (Fig. 6), it induces a slight membrane shrinkage and pore reduction.
In order to investigate the light responsive behaviour of this membrane in terms of performance, water flux and protein retention measurements were carried out for the irradiated and non-irradiated membranes. The water flux (Fig. 8) for the PS44k-b-PAnMMA1.2k-b-PMMA45k membrane was 270 L m−2 h−1 bar−1 and for the PS43k-b-PAnMMA1.5k-b-PMMA105k membrane was 310 L m−2 h−1 bar−1. The membranes irradiated for 24 h at 365 nm showed a decrease in water flux. This was observed for both copolymers. The water flux for the photo-dimerized membrane went down to 230 L m−2 h−1 bar−1 for PS44k-b-PAnMMA1.2k-b-PMMA45k and 250 L m−2 h−1 bar−1 for PS43k-b-PAnMMA1.5k-b-PMMA105k. To check the reversibility, the photo-dimerized membranes were then irradiated at 264 nm for 6 h. The water flux recovered to 250 L m−2 h−1 bar−1 for PS44k-b-PAnMMA1.2k-b-PMMA45k and 280 L m−2 h−1 bar−1 for PS43k-b-PAnMMA1.5k-b-PMMA105k, after 264 nm irradiation. The membrane retention was measured using two different model proteins, bovine serum albumin (BSA 66 kg mol−1) and γ-globulin (IgG, 150 kg mol−1). The rejection measurements were carried out and the concentrations of proteins were measured using UV visible spectroscopy. Both membranes (Fig. 8) rejected more than 95% γ-globulin. The rejection of BSA was 70% and 62% respectively. Irradiation at 365 nm for 24 h slightly increased the membranes rejection of BSA to 75% (low molecular weight) and 68% (high molecular weight). A significant chemical change in the anthracene block was observed from the fluorescence spectra. The membrane flux and retention changed in response to light, but the changes were small due to the relatively short anthracene segment, which is the light active block. Larger blocks would be more effective if larger effects on flux and selectivity would be targeted.
 | ||
| Fig. 8 Water flux and retention measurements. | ||
Conclusions
In summary, we successfully developed nanostructured triblock copolymer membranes with photoresponsive behavior by non-solvent induced phase separation. Hexagonally order or cylindrical pores were obtained, depending on the molecular weight used. The membranes are responsive to light. Their flux and retention were altered on irradiating at longer (365 nm) and shorter wavelength (264 nm). The anthracene moiety present in PS-b-PAnMMA-b-PMMA block copolymer membranes, on exposure to UV at 365 nm, converted into cyclic adduct in the solid state. Then the cyclic adduct was reversibly disrupted on irradiation with shorter wavelength of light (264 nm).Acknowledgements
This work was supported by the King Abdullah University of Science and Technology (KAUST), in the frame of a CRG2 grant.References
- S. Darvishmanesh, X. Qian and S. R. Wickramasinghe, Curr. Opin. Chem. Eng., 2015, 8, 98–104 CrossRef.
- F. P. Nicoletta, D. Cupelli, P. Formoso, G. De Filpo, V. Colella and A. Gugliuzza, Membranes, 2012, 2, 134–197 CrossRef CAS PubMed.
- S. J. Lue, J.-J. Hsu, C.-H. Chen and B.-C. Chen, J. Membr. Sci., 2007, 301, 142–150 CrossRef CAS.
- Q. Yang, H. H. Himstedt, M. Ulbricht, X. Qian and S. R. Wickramasinghe, J. Membr. Sci., 2013, 430, 70–78 CrossRef CAS.
- L. Baumann, K. Schöller, D. de Courten, D. Marti, M. Frenz, M. Wolf, R. M. Rossi and L. J. Scherer, RSC Adv., 2013, 3, 23317–23326 RSC.
- D. Wandera, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2010, 357, 6–35 CrossRef CAS.
- Z. Liu, W. Wang, R. Xie, X.-J. Ju and L.-Y. Chu, Chem. Soc. Rev., 2016, 45, 460–475 RSC.
- C. Zhao, S. Nie, M. Tang and S. Sun, Prog. Polym. Sci., 2011, 36, 1499–1520 CrossRef CAS.
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Nat. Mater., 2005, 4, 298–302 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- I. Tokarev and S. Minko, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- H. H. Himstedt, H. Du, K. M. Marshall, S. R. Wickramasinghe and X. Qian, Ind. Eng. Chem. Res., 2013, 52, 9259–9269 CrossRef CAS.
- N. Liu, D. R. Dunphy, P. Atanassov, S. D. Bunge, Z. Chen, G. P. López, T. J. Boyle and C. J. Brinker, Nano Lett., 2004, 4, 551–554 CrossRef CAS.
- L. Y. Chu, Y. Li, J. H. Zhu and W. M. Chen, Angew. Chem., Int. Ed., 2005, 44, 2124–2127 CrossRef CAS PubMed.
- D. He, H. Susanto and M. Ulbricht, Prog. Polym. Sci., 2009, 34, 62–98 CrossRef CAS.
- L.-Y. Chu, Y. Li, J.-H. Zhu, H.-D. Wang and Y.-J. Liang, J. Controlled Release, 2004, 97, 43–53 CrossRef CAS PubMed.
- S. P. Nunes, M. Karunakaran, N. Pradeep, A. R. Behzad, B. Hooghan, R. Sougrat, H. He and K.-V. Peinemann, Langmuir, 2011, 27, 10184–10190 CrossRef CAS PubMed.
- S. P. Nunes, A. R. Behzad, B. Hooghan, R. Sougrat, M. Karunakaran, N. Pradeep, U. Vainio and K.-V. Peinemann, ACS Nano, 2011, 5, 3516–3522 CrossRef CAS PubMed.
- F. Schacher, M. Ulbricht and A. H. Müller, Adv. Funct. Mater., 2009, 19, 1040–1045 CrossRef CAS.
- W. A. Phillip, R. Mika Dorin, J. R. Werner, E. M. Hoek, U. Wiesner and M. Elimelech, Nano Lett., 2011, 11, 2892–2900 CrossRef CAS PubMed.
- R. Shevate, M. Karunakaran, M. Kumar and K.-V. Peinemann, J. Membr. Sci., 2016, 501, 161–168 CrossRef CAS.
- Y. Kim, S. Kwon, D. Yoo, M. F. Rubner and M. S. Wrighton, Chem. Mater., 1997, 9, 2699–2701 CrossRef CAS.
- K. Nakabayashi, S. Inoue, Y. Abiko and H. Mori, Macromolecules, 2013, 46, 4790–4798 CrossRef CAS.
- H. Zhang, W. Verboom and D. N. Reinhoudt, Tetrahedron Lett., 2001, 42, 4413–4416 CrossRef CAS.
- K. S. Wei and R. Livingston, Photochem. Photobiol., 1967, 6, 229–232 CrossRef CAS.
- M. M. Maturi, G. Fukuhara, K. Tanaka, Y. Kawanami, T. Mori, Y. Inoue and T. Bach, Chem. Commun., 2016, 52, 1032–1035 RSC.
- G. W. Breton and X. Vang, J. Chem. Educ., 1998, 75, 81 CrossRef CAS.
- Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228–5234 CrossRef CAS.
- K. Rameshbabu, Y. Kim, T. Kwon, J. Yoo and E. Kim, Tetrahedron Lett., 2007, 48, 4755–4760 CrossRef CAS.
- C. Wang, D. Zhang, J. Xiang and D. Zhu, Langmuir, 2007, 23, 9195–9200 CrossRef CAS PubMed.
- J. S. Trent, Macromolecules, 1984, 17, 2930–2931 CrossRef CAS.
- M. Karunakaran, S. P. Nunes, X. Qiu, H. Yu and K.-V. Peinemann, J. Membr. Sci., 2014, 453, 471–477 CrossRef CAS.
- S. P. Nunes, Macromolecules, 2016, 49, 2905–2916 CrossRef CAS.
- S. P. Gido, D. W. Schwark, E. L. Thomas and M. do Carmo Goncalves, Macromolecules, 1993, 26, 2636–2640 CrossRef CAS.
- A. Knoll, A. Horvat, K. Lyakhova, G. Krausch, G. Sevink, A. Zvelindovsky and R. Magerle, Phys. Rev. Lett., 2002, 89, 035501 CrossRef CAS PubMed.
- A. Knoll, R. Magerle and G. Krausch, J. Chem. Phys., 2004, 120, 1105–1116 CrossRef CAS PubMed.
- A. Horvat, A. Knoll, G. Krausch, L. Tsarkova, K. Lyakhova, G. Sevink, A. Zvelindovsky and R. Magerle, Macromolecules, 2007, 40, 6930–6939 CrossRef CAS.
- S. Ludwigs, A. Böker, A. Voronov, N. Rehse, R. Magerle and G. Krausch, Nat. Mater., 2003, 2, 744–747 CrossRef CAS PubMed.
- I. Hamley, Prog. Polym. Sci., 2009, 34, 1161–1210 CrossRef CAS.
- J. N. Albert and T. H. Epps, Mater. Today, 2010, 13, 24–33 CrossRef CAS.
- J. You, Y. Kim and E. Kim, Mol. Cryst. Liq. Cryst., 2010, 520, 128/[404]–135/[411] Search PubMed.
- I. Zouev, D.-K. Cao, T. Sreevidya, M. Telzhensky, M. Botoshansky and M. Kaftory, CrystEngComm, 2011, 13, 4376–4381 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Solubility parameter and membrane morphologies are provided. See DOI: 10.1039/c6ra14317b |
| This journal is © The Royal Society of Chemistry 2016 |