
Hexaphenylbenzene and hexabenzocoronene-based porous polymers for the adsorption of volatile organic compounds†
Abstract
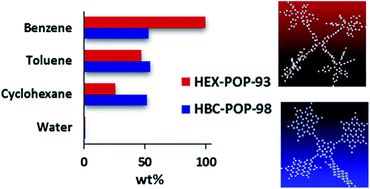
Toxic volatile organic compounds (VOCs) including benzene and toluene that are emitted from industrial chemical processes and outdoor or indoor chemical applications are harmful to the environment and threaten human health. Porous organic polymers (POPs), which have attracted much interest during the last decade as materials for gas adsorption and separation, have only recently gained attention for the adsorption of VOCs. We synthesised two POPs based on hexaphenylbenzene (HEX-POP-93) and hexabenzocoronene (HBC-POP-98) and studied the adsorption of organic vapours. Interestingly, while both POPs have moderate BET surface areas (687 and 548 m2 g−1 respectively), they both show an excellent affinity for organic vapours over water, with a high benzene adsorption capacity of 99.9 wt% for HEX-POP-93.
 Please wait while we load your content...
Please wait while we load your content...

