
FeF3-catalyzed MCR in PEG-400: ultrasound assisted synthesis of N-substituted 2-aminopyridines†
Dinne Naresh Kumar Reddyab,
Kothapalli Bannoth Chandrasekharb,
Yaddanapudi Sesha Siva Ganesha,
G. Rajeshwar Reddya,
J. Pradeep Kumara,
Ravi Kumar Kapavarapuc and
Manojit Pal*d
aCustom Pharmaceuticals Services and Dr Reddy's Laboratories Ltd, Bollaram Road, Miyapur, Hyderabad 500049, India
bCollege of Engineering, JNTU, Anantapur 533 003, Andhra Pradesh, India
cDoctoral Programme in Experimental Biology and Biomedicine, Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal
dDr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500046, India. E-mail: manojitpal@rediffmail.com
First published on 8th July 2016
Abstract
FeF3 catalyzed four component reaction under ultrasound irradiation was explored for the first time to prepare N-substituted 2-aminopyridines in good yields. The methodology involved the use of readily available starting materials and PEG-400 under mild reaction conditions in the presence of air. The one-pot methodology afforded a range of compounds of pharmacological interest indicating its potential in generating a diversity based library of small molecules useful for medicinal chemistry and drug discovery.
Being a well known N-heterocycle in many bioactive compounds and natural products1 the pyridine ring is considered as one of the privileged structures in medicinal chemistry and drug discovery. For example, the pyridine framework has been explored for the discovery and development of several marketed drugs e.g. etoricoxib2 (a cyclooxygenase-2 inhibitor, Fig. 1), piclamilast and roflumilast [inhibitors of phosphodiesterase 4 (PDE4)] (Fig. 1). The PDE4 inhibitors are known to be beneficial for the potential treatment of asthma and chronic obstructive pulmonary disease (COPD).3a To date roflumilast (Daxas® by Nycomed and Daliresp by Forest Lab)3b and apremilast (Otezla by Celgene) have been launched in Europe and US. However, adverse side effects such as nausea, emesis and cardiovascular issues have halted the progress of several other PDE4 inhibitors. Thus the search for a new chemical class possessing PDE4 inhibitory properties with improved therapeutic indexes is desirable. Accordingly, we have explored the 4-aryl substituted cyano pyridines (A, Fig. 1) for this purpose.4 In further continuation of this research we became interested in evaluating PDE4 inhibitory properties of some N-substituted 2-amino analogs of A (e.g. B, Fig. 1) possessing an alkyl/aryl/heteroaryl moiety at the C-4 position of the pyridine ring. Our major focus was on the C-4 substituent as this group was found to be crucial for activity in the case of piclamilast and roflumilast (Fig. 1). We therefore required a robust and efficient method to generate a library of compounds based on the template B having a range of groups at the C-4 position for the detailed Structure Activity Relationship (SAR) studies.
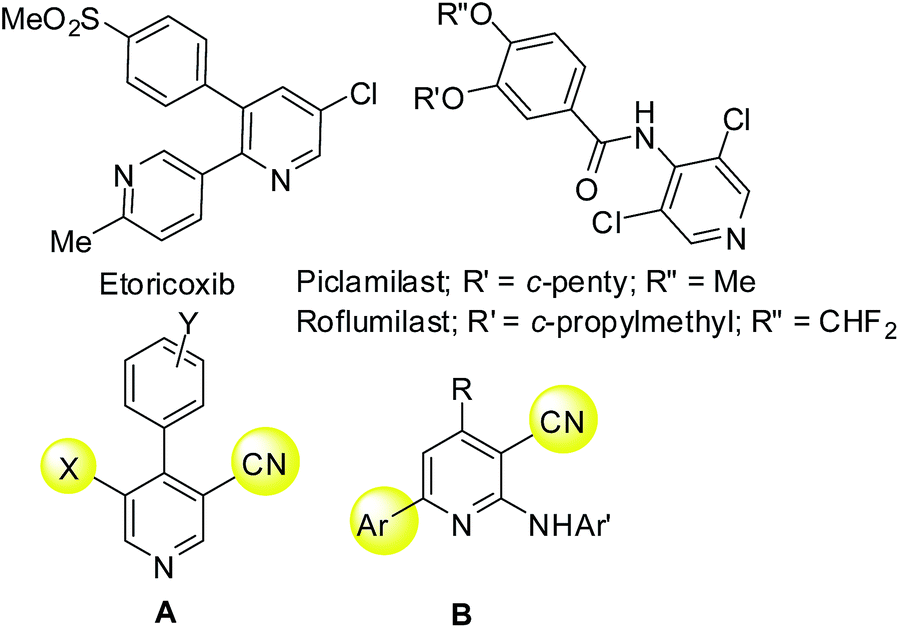 | ||
| Fig. 1 Pyridine based known PDE4 inhibitors and design of new inhibitors (B). | ||
In view of their usefulness in organic synthesis5 several syntheses of 2-aminopyridine derivatives6–9 have been reported that generally involves the reaction of 2-halo or 2-alkoxypyridines with ammonia or amines. The use of multi-component reactions (MCR) has also been explored for the synthesis of this class of compounds.8,9 Indeed, a three component reaction involving the reaction of α,β-unsaturated ketones, cyano derivatives and amines afforded the desired pyridine derivatives.8 Later a modified synthesis of 2-aminopyridines was reported via the reaction of α,β-unsaturated ketones with malononitrile and amines or ammonium acetate under mild conditions.9 However, due to the several drawbacks associated with these previously reported methods including poor to moderate yields, longer reaction time and limited substrate scope a faster method has been developed recently. In this strategy the MCR of α,β-unsaturated ketones (chalcones), malononitrile and amines was accelerated by microwave irradiation to achieve a chemoselective synthesis of N-substituted 2-aminopyridines.10 Indeed, this method appeared to be an effective, faster (3–9 min) and good yielding (76–90%) process though it involved the use of DMF as a solvent and chalcone as one of the reactants. In another approach, following our earlier synthesis of pyridine derivatives via a 4-component reaction of β-ketoester, arylaldehyde, malononitrile and an alcohol4 a FeCl3-catalyzed similar 4-component reaction using arylamine in place of alcohol was developed for the synthesis of N-substituted 2-aminopyridines in 38–86% yield.11a Subsequently, a “Sn” mediated MCR in water leading to the same class of pyridines was reported by us.11b Once again all these methods involved relatively longer reaction time. The ultrasound assisted organic reactions12 have attracted enormous attention due to the efficiency (e.g. shorter reaction time, milder conditions, higher yields etc.) and greenness (in terms of energy conservation and waste minimization) of these processes over the conventional heating methods. On the other hand, because of its non-hazardous nature polyethyleneglycol 400 (PEG-400) is considered as an environmental friendly solvent in various organic reactions.13 Thus, in search of a more convenient and straightforward method for the synthesis of N-substituted 2-aminopyridines we decided to explore the use of ultrasound and PEG-400 for our purpose. Indeed, we were successful in our effort. Herein, we report a FeF3 mediated four component reaction (4-CR) under ultrasound irradiation leading to the target pyridine derivatives from readily available starting materials (Scheme 1). Though as a catalyst FeF3 has received some attention in organic synthesis14,15 its use in the synthesis of pyridines has not been explored. To the best of our knowledge this is the first use of ultrasound assisted FeF3-catalyzed MCR for the synthesis of this class of compounds.
 | ||
| Scheme 1 Ultrasound assisted 4-component reaction leading to N-substituted 2-aminopyridines (5). | ||
In the beginning of our study, it was necessary to test the feasibility of our anticipated ultrasound based 4-component reaction leading to the formation of the desired pyridine ring. Accordingly, the commercially available acetophenone (1a), benzaldehyde (2a), o-toluidine (3a) and malononitrile (4) were chosen as reactants for the MCR that was performed under various conditions in open air (Table 1). Initially, we performed the reaction in water in the presence of a range of catalysts under thermal heating (at 80 °C) for 10–11 h (entries 1–10, Table 1). However, except SnCl2·2H2O and FeF3 none of other catalyst was found to be effective under the condition employed. We then opted for another green solvent i.e. PEG-400 (entries 11–16, Table 1). Once again SnCl2·2H2O and FeF3 were found to be best among all the catalysts tested when the desired product 5a was isolated in good yields. In order to decrease the reaction time and temperature we performed the reaction catalyzed by SnCl2·2H2O or FeF3 under ultrasound irradiation in both water and PEG-400, respectively (entries 17–20, Table 1). To our delight the reaction was completed within 3 h in these cases when performed at 60 °C affording the product 5a in 80–92% yield. Since the best yield of 5a was obtained under the condition of entry 20 of Table 1 in the presence of FeF3 hence this was identified as optimal reaction condition. The role of solvent PEG-400 and catalyst FeF3 as well as effect of temperature at lower and higher than 60 °C was also examined (entries 21–25, Table 1). While the reaction proceeded under the solvent free condition (entry 21, Table 1) almost no product was formed in the absence of catalyst (entry 22, Table 1). The use of higher reaction temperature did not improve the product yield further (entry 23, Table 1) whereas a decrease in temperature lowered the yield (entry 24 and 25, Table 1). To optimize the catalyst loading the reaction was performed in the presence of 5, 10, 15 and 20 mol% of FeF3 when 5a was obtained in 70, 92, 93 and 90% yield respectively indicating 10 mol% of catalyst as optimal amount. Notably, the reaction afforded 5a in 30% yield along with 2-(3-oxo-1,3-diphenylpropyl)malononitrile (6) as a major product (60% yield) when stopped after 30 min (entry 26, Table 1). This observation suggested that the compound 6 is an intermediate in the present reaction. Finally, we examined the use of FeCl3 as a catalyst in place of FeF3 (entry 27, Table 1). While the reaction proceeded well in this case affording 5a in good yield the duration of the reaction was 6 h.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a All the reactions were performed using 1a (1.25 mmol), 2a (1.25 mmol), 3a (1.25 mmol), 4 (1.25 mmol), and a catalyst (10 mol%) in a solvent (0.3 mL) under open air.b Isolated yield.c The reaction was performed under ultrasound irradiation.d The compound 2-(3-oxo-1,3-diphenylpropyl) malononitrile (6) was isolated as a major product (60% yield) in this case. | |||||
| 1 | PMA–SiO2 | H2O | 80 | 10 | Traces |
| 2 | SiO2 | H2O | 80 | 10 | Traces |
| 3 | Clay | H2O | 80 | 10 | 17 |
| 4 | CeCl3·7H2O | H2O | 80 | 10 | 30 |
| 5 | Citric acid | H2O | 80 | 10 | 25 |
| 6 | PTSA | H2O | 80 | 10 | 30 |
| 7 | L-Proline | H2O | 80 | 10 | Traces |
| 8 | BiCl3 | H2O | 80 | 11 | 40 |
| 9 | SnCl2·2H2O | H2O | 80 | 10 | 75 |
| 10 | FeF3 | H2O | 80 | 10 | 80 |
| 11 | PMA–SiO2 | PEG-400 | 80 | 10 | 20 |
| 12 | Amberlite | PEG-400 | 80 | 10 | Traces |
| 13 | Indion resin | PEG-400 | 80 | 10 | Traces |
| 14 | PTSA | PEG-400 | 80 | 10 | 20 |
| 15 | SnCl2·2H2O | PEG-400 | 80 | 10 | 70 |
| 16 | FeF3 | PEG-400 | 80 | 10 | 85 |
| 17 | SnCl2·2H2O | H2O | 60 | 3 | 80c |
| 18 | FeF3 | H2O | 60 | 3 | 80c |
| 19 | SnCl2·2H2O | PEG-400 | 60 | 3 | 81c |
| 20 | FeF3 | PEG-400 | 60 | 3 | 92c |
| 21 | FeF3 | No solvent | 60 | 3 | 60c |
| 22 | No catalyst | PEG-400 | 60 | 3 | Tracesc |
| 23 | FeF3 | PEG-400 | 70 | 3 | 91c |
| 24 | FeF3 | PEG-400 | 50 | 3 | 60c |
| 25 | FeF3 | PEG-400 | RT | 3 | 40c |
| 26 | FeF3 | PEG-400 | 60 | 0.5 | 30d |
| 27 | FeCl3 | PEG-400 | 60 | 6 | 80 |

Having identified the best reaction conditions we focused on assessing the generality and scope of this methodology. Accordingly, a variety of compounds were prepared by using this FeF3 catalyzed methodology (Table 2). A range of aldehydes (2) containing alkyl, aryl and heteroaryl moieties were employed. Indeed, alkyl moiety may include propyl (linear) or s-butyl (branched) group whereas the aryl ring may contain a strong electron donating (e.g. OMe) or electron withdrawing (e.g. NO2) group. The heteroaryl moiety may include thienyl or furan group. The aryl ring of ketone (1) may bear substituent like OMe, Cl or CN whereas a range of substituents like Me, F, Br and CN may present on the benzene ring of aniline derivatives (3) employed. The reaction proceeded well in all these cases affording the corresponding pyridine derivatives in good to excellent yields. Notably, these reactions do not require the use of any inert or anhydrous atmosphere. The use of an aliphatic amine i.e. n-BuNH2 was also explored. However the reaction afforded a 2,6-dicyanoaniline derivative10 (7) instead of the desired 2-aminopyridine derivative. All the compounds synthesized were characterized by spectral and analytical data. For example, the IR absorption in the range 2220–2210 cm−1 indicated the presence of CN moiety. This was further supported by the appearance of a 13C signal near 110 ppm (due to the CN moiety) and 90 ppm (due to the C-4 i.e. the CN bearing carbon of the pyridine ring) in the 13CNMR spectra. Further a singlet appeared near δ 7.30 (though it was merged with other signals in some cases) in the 1HNMR spectra were due to the C-5 proton of the pyridine ring.


Based on the results presented in Table 1 especially the isolation of compound 6, a plausible reaction mechanism is proposed in Scheme 2. Thus, the intermediate E-1 is formed16 in situ via the Knoevenagel condensation between 2 and 4 assisted by ultrasound. The catalyst FeF3 appeared to play the role of a Lewis acid in the present reaction.14a Consequently, coordination of the nitrogen lone pair of E-1 with the FeF3 facilitates the Michael type of reaction17 with the enol form of 1 affording the intermediate E-2 (cf. compound 6, entry 26 of Table 1). A further coordination of nitrogen lone pair of E-2 with the FeF3 (path a) facilitates a nucleophilic attack by the arylamine 3 followed by isomerization of the resulting species to give the intermediate E-3. An intramolecular cycloaddition of E-3 (via the loss of a water molecule) followed by oxidative aromatization in the presence of air afforded the desired product 5. The E-2 seems to follow a different pathway (path b) when an aliphatic amine i.e. n-BuNH2 was used. Being a stronger base than aromatic amines, n-BuNH2 facilitated Knoevenagel condensation of E-2 with 4 leading to E-4 which on subsequent intramolecular cyclization, HCN elimination and aromatization afforded 7.10
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
The results of Table 1 (entry 16 vs. 20) clearly suggest that the synthesis of compound 5 was accelerated in the presence of ultrasound. It is known that cavitation caused by ultrasound is involved with the growth, oscillation, and collapse of bubbles under the action of an acoustic field.18 The cavitational collapse on the other hand creates drastic conditions (e.g. the temperature of 2000–5000 K and pressure up to 1800 atmosphere) inside the medium within an extremely short period of time. Also, strong physical effects including shear forces, jets, and shock waves are caused by this collapse outside the bubble. Thus, these cavitation-induced overall effects perhaps explains the rate acceleration of the present reaction under ultrasound in the absence of which the reaction took relatively longer time and higher temperature (entry 16, Table 1).19 Nevertheless, to gain further evidence, 2-(4-methoxybenzylidene)malononitrile (8) was prepared16a from 4-methoxybenzaldehyde (2b) and malononitrile (4) separately and treated with acetophenone (1a) and 4-fluoroaniline (3d) under the conditions of entry 20 of Table 1. The isolation of pyridine derivative 5d in 90% yield (Scheme 3) suggested intermediacy of E-1 (Scheme 2) in the present reaction.
 | ||
| Scheme 3 The reaction of intermediate 8 with 1a and 3d. | ||
To assess the PDE4 inhibitory potential of this class of compounds some of the pyridines synthesized were tested in vitro at 10 μM using PDE4B enzyme assay20a along with rolipram20b as a reference compound. Accordingly, compound 5d and 5j showed 69.0 ± 1.21 and 65.0 ± 2.04% inhibition compared to rolipram's 90 ± 4.30% inhibition whereas 5i did not show any inhibition. Rest of the compounds showed 40–60% inhibition. It is evident that the nature of substituent(s) present on the C-4 aryl moiety seemed to have significant influence on PDE4 activities and the strong electron donating group OMe at the para position of the C-4 benzene ring was found to be favorable. This was further supported by the in silico docking studies performed by compound 5d (Fig. 2) and 5j (see ESI†) where the OMe group participated in favorable hydrophobic interactions with PDE4B.21 Further in vitro studies are ongoing using these compounds.
 | ||
| Fig. 2 Docking of compound 5d into PDE4B: dotted white bond showing H-bond interactions and yellow bonds shows hydrophobic interactions with the binding site residues, protein is represented by sticks and is colored according to the atom types. | ||
In conclusion, we have developed an ultrasound assisted four component reaction for the synthesis of N-substituted 2-aminopyridines. The combination of catalyst FeF3 and PEG-400 was found to be most effective for this MCR. The advantages and drawbacks of the methodology are presented. The methodology is free from the use of inert or anhydrous atmosphere and involved the use of commercially available starting materials e.g. acetophenones, arylaldehydes, anilines and malononitrile to give the desired pyridine derivatives under mild reaction conditions. A range of N-substituted 2-aminopyridines were prepared by using this one-pot methodology in good to excellent yields (85–92%) within 3 h. Two of these compounds showed encouraging PDE4 inhibition in vitro and interaction in silico. Though as a catalyst FeF3 has received some attention in organic synthesis however its uses are not only uncommon in the synthesis of pyridines but also in MCR. Additionally, our study highlights the potential of the present FeF3 catalyzed methodology in generating diversity based library of small molecules related to 2-aminopyridine useful for medicinal/pharmaceutical chemistry and drug discovery. Thus the present methodology may attract considerable attention both in organic synthesis and medicinal chemistry.
Acknowledgements
D. N. K. Reddy thanks Dr P Mahesh Kumar, Dr M. Srinivasa Rao, Dr H. Ramamohan and the management of CPS, DRL, Hyderabad, India for support and encouragements (clearence no: IPDOIPM-00508). Authors thank Dr K. Parsa and his group of DRILS, Hyderabad for PDE4 assay.Notes and references
- (a) Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, 1996, vol. 5 Search PubMed; (c) J. A. Varela and C. Sáa, Chem. Rev., 2003, 103, 3787 CrossRef CAS PubMed; (d) A. R. Katritzky, Chem. Rev., 2004, 104, 2125 CrossRef CAS.
- A. K. Matsumoto and P. F. Cavanaugh Jr, Drugs Today, 2004, 40, 395 CrossRef CAS PubMed.
- (a) A. Kodimuthali, S. S. L. Jabaris and M. Pal, J. Med. Chem., 2008, 51, 5471 CrossRef CAS PubMed; (b) D. Price, A. Chisholm, D. Ryan, A. Crockett and R. Jones, Prim. Care Respir. J., 2010, 19, 342 CrossRef PubMed.
- T. R. Reddy, G. R. Reddy, L. S. Reddy, S. Jammula, Y. Lingappa, R. Kapavarapu, C. L. T. Meda, K. V. L. Parsa and M. Pal, Eur. J. Med. Chem., 2012, 48, 265 CrossRef PubMed.
- (a) K. Pericherla, P. Kaswan, K. Pandey and A. Kumar, Synthesis, 2015, 47, 887 CrossRef CAS; (b) A. K. Bagdi and A. Hajra, Chem. Rec., 2016 DOI:10.1002/tcr.201600057.
- (a) K. L. Howard, H. W. Stewart, E. A. Conroy and J. J. Denton, J. Org. Chem., 1953, 18, 1484 CrossRef CAS; (b) K. Matsumoto, H. Minatogawa, M. Munakata, M. Toda and H. Tsukube, Tetrahedron Lett., 1990, 27, 3923 Search PubMed; (c) K. Matsumoto, S. Hashimoto and S. Otani, J. Chem. Soc., Chem. Commun., 1991, 306 RSC; (d) S. V. Tolkunov, M. N. Kalnitskii and E. A. Zemskaya, Chem. Heterocycl. Compd., 1992, 27, 1253 CrossRef; (e) H. Vorbruggen, Adv. Heterocycl. Chem., 1990, 49, 117 CrossRef; (f) J. M. Quintela, C. Peinador, M. C. Veiga, L. M. Botana, A. Alfonso and R. Riguere, Eur. J. Med. Chem., 1998, 33, 887 CrossRef CAS.
- A. A. M. Abdel-Aziz, H. I. El-Subbagh and T. Kunieda, Bioorg. Med. Chem., 2005, 13, 4929 CrossRef CAS PubMed.
- A. R. Katritzky, S. A. Belyakov, A. E. Sorochinsky, S. A. Henderson and J. Chen, J. Org. Chem., 1997, 62, 6210 CrossRef CAS.
- (a) M. A. I. Salem, H. M. F. Madkour, E. S. A. Soliman and N. F. H. Mahmoud, Heterocycles, 2000, 53, 1129 CrossRef CAS; (b) V. Raghukumar, D. Thirumalai, V. T. Ramakrishnan, V. Karunakara and P. Ramamurthy, Tetrahedron, 2003, 59, 3761 CrossRef CAS.
- S. Tu, B. Jiang, Y. Zhang, R. Jia, J. Zhang, C. Yao and F. Shi, Org. Biomol. Chem., 2007, 5, 355 CAS.
- (a) X. He, Y. Shang, Z. Yu, M. Fang, Y. Zhou, G. Han and F. Wu, J. Org. Chem., 2014, 79, 8882 CrossRef CAS PubMed; (b) D. N. K. Reddy, K. B. Chandrasekhar, Y. S. S. Ganesh, B. S. Kumar, R. Adepu and M. Pal, Tetrahedron Lett., 2015, 56, 4586 CrossRef CAS.
- (a) G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180 RSC; (b) J. T. Li, S. X. Wang, G. F. Chen and T. S. Li, Curr. Org. Synth., 2005, 2, 415 CrossRef CAS; (c) N. Ratoarinoro, A. M. Wilhelm, J. Berlan and H. Delmas, Chem. Eng. J., 1992, 50, 27 CrossRef CAS.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64 RSC.
- (a) B. P. Bandgar and V. T. Kamble, Green Chem., 2001, 3, 265 RSC; (b) T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2007, 32, 9844 CrossRef PubMed; (c) V. T. Kamble, B. P. Bandgar, S. B. G. Suryawanshi and S. N. Bavikar, Aust. J. Chem., 2006, 59, 837 CrossRef CAS; (d) R. Surasani, D. Kalita, A. V. D. Rao, K. Yarbagi and K. B. Chandrasekhar, J. Fluorine Chem., 2012, 135, 91 CrossRef CAS; (e) B. P. Bandgar, V. T. Kamble and A. Kulkarni, Aust. J. Chem., 2005, 58, 607 CrossRef CAS; (f) X.-L. Fang, R.-Y. Tang, X.-G. Zhang and J.-H. Li, Synthesis, 2011, 1099 CAS.
- T. B. Kumar, C. Sumanth, A. V. D. Rao, D. Kalita, M. S. Rao, K. B. C. Sekhar, K. S. Kumar and M. Pal, RSC Adv., 2012, 2, 11510 RSC.
- (a) See for example: F. Bigi, M. L. Conforti, R. Maggi, A. Piccinno and G. Sartori, Green Chem., 2000, 2, 101 RSC; (b) For synthesis of pyridine derivatives from in situ generated benzylidenemalononitriles, see: M. N. Khan, S. Pal, S. Karamthulla and L. H. Choudhury, RSC Adv., 2014, 4, 3732 RSC; (c) Notably, omission of ketone 1 from this 4-component reaction i.e. the reaction of benzaldehyde, aniline and malononitrile under the condition of entry 20 of Table 1 afforded 2-amino-4-phenylquinoline-3-carbonitrile (cf. A. Khalafi-Nezhad, S. Sarikhani, E. S. Shahidzadeh and F. Panahi, Green Chem., 2012, 14, 2876 RSC ) formation of which can also be explained by the in situ generation of E-1 type intermediate (Scheme 2).
- For Michael addition of a carbonyl compound to benzylidenemalonitriles, see: C. W. Suh and D. Y. Kim, Bull. Korean Chem. Soc., 2014, 35, 98, DOI:10.5012/bkcs.2014.35.1.98.
- (a) T. J. Mason and D. Peters, Practical Sonochemistry, Ellis Horwood, New York, NY, USA, 1991 Search PubMed; (b) T. J. Mason, Ultrason. Sonochem., 2007, 14, 476 CrossRef CAS PubMed.
- To test the scalability of this MCR and recyclability of the catalyst the reaction was performed at a gm scale [e.g. acetophenone (1a, 4.0 g, 0.033 mmol), 4-nitrobenzaldehyde (2e, 5.03 g, 0.033 mmol), aniline (3b, 3.1 g, 0.033 mmol), malononitrile 4, 2.2 g, 0.033 mmol and FeF3 (0.375 g, 10 mol%) in PEG-400 (8 mL)] when the desired product 5i was isolated in 90% yield (see ESI†). The filtered catalyst was collected, dried under vacuum below 50 °C and reused for next cycle of the same reaction when 5i was isolated in 86% yield.
- (a) P. Wang, J. G. Myers, P. Wu, B. Cheewatrakoolpong, R. W. Egan and M. M. Billah, Biochem. Biophys. Res. Commun., 1997, 234, 320 CrossRef CAS PubMed; (b) J. Demnitz, L. LaVecchia, E. Bacher, T. H. Keller, T. Muller, F. Schurch, H. P. Weber and E. Pombo-Villar, Molecules, 1998, 3, 107 CrossRef CAS.
- (a) The molecular docking simulations were carried out using GRIP method of docking in Biopredicta module of Vlife MDS (Molecular Design Suite) 4.6 (for detailed procedure, see ESI†); (b) G. L. Card, B. P. England, Y. Suzuki, D. Fong, B. Powell, B. Lee, C. Luu, M. Tabrizizad, S. Gillette, P. N. Ibrahim, D. R. Artis, G. Bollag, M. V. Milburn, S. H. Kim, J. Schlessinger and K. Y. Zhang, Structure, 2004, 12, 2233 CrossRef CAS PubMed; (c) A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of the 1H and 13C NMR spectra, HRMS. See DOI: 10.1039/c6ra14228a |
| This journal is © The Royal Society of Chemistry 2016 |