
A spiro-type ammonium based switchable dielectric material with two sequential reversible phase transitions above room temperature†
Abstract
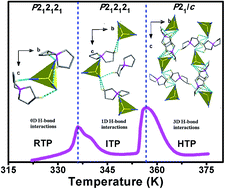
An organic–inorganic hybrid compound 5-azonia-spiro[4,4]nonane tetrabromocadmium (1, [ASN]2[CdBr4], ASN = (CH2)4N(CH2)4), has been discovered as a new phase transition material. Differential scanning calorimetry (DSC) and dielectric measurements reveal that 1 undergoes dielectric anomalies which could be tuned in three evident dielectric states and switched by two sequential reversible phase transitions around 336 and 357 K, respectively. Detailed variable-temperature single-crystal structural analyses indicate that the distinct twisting motions of the flexible [ASN]+ ammonium cationic moieties and the relative reorientations of both the ions contribute to the structure phase transitions of 1 triggered by temperature. Particularly, 1 displays a switchable SHG response in the vicinity of 357 K, where the temperature-dependent dielectric permittivity of 1 changes abruptly from 8 to 15, with a large thermal hysteresis of 19 K. Therefore, such a distinctive dielectric performance discloses that 1 might be an interesting high-temperature switchable dielectric and nonlinear optical material. All these results open a new venue to design novel phase transition materials through selecting the flexible spiro-type ammonium salts.
 Please wait while we load your content...
Please wait while we load your content...

