DOI:
10.1039/C6RA14070J
(Paper)
RSC Adv., 2016,
6, 104897-104910
Efficient batch and column removal of Cr(VI) by carbon beads with developed nano-network†
Received
30th May 2016
, Accepted 20th October 2016
First published on 21st October 2016
Abstract
Chromium removal is of significance to our daily life; however, the fabrication of high-performance sorbents with practicality remains a challenge. Herein, alginate-derived carbon beads (Alg-CB-X) with a developed nano-network were successfully prepared through a simple carbothermal reduction at different carbonization temperatures and acid treatment. The aim of this material was for easy separation and removal of Cr(VI) from water. The materials were characterized by FESEM, TEM, XRD, FTIR, XPS, EDS, Raman spectrometry and N2 sorption measurements. Parameters affecting the uptake of Cr(VI) ions by Alg-CB-X such as solution pH, initial concentration, contact time and coexisting anions were investigated systematically in batch experiments. The mechanism investigation showed that the removal of Cr(VI) involved electrostatic attraction and slightly involved redox reactions. However, it is emphasized that the reduction of Cr(VI) to Cr(III) was very slow and occurred only slightly because of the lack of oxygen-containing functional groups (caused by the pyrolysis process) and the short adsorption period. With increasing experiment time and initial concentration of Cr(VI), the amount of reduced Cr(III) increased gradually. Finally, Cr(VI) is deposited onto the carbon surface, and the formed Cr(III) is released into the aqueous phase due to electronic repulsion between the positively-charged groups and the Cr(III).
1. Introduction
Nowadays, the wide spread use of chemical compounds and hazardous materials around the world has led to serious water pollution. Heavy metal ions are one of the most dangerous water pollutants. They can form compounds within the bio-system that have a strong tendency to be potentially carcinogenic and mutagenic; and they may lead to the lethal poisoning of human beings and animals, even at ultralow concentrations.1–3 One such hazardous heavy metal is chromium. It is reported that chromium, coming from leather tanning, mining operations, electroplating, pigment manufacturing and so on, is the second most abundant inorganic groundwater contaminant at hazardous waste sites.4–6 In natural water, chromium is present as both Cr(III) and Cr(VI). By contrast, Cr(III) is an essential micronutrient that helps the body in metabolizing sugar, protein and fat (requirement is 50–200 mg per day). However, Cr(VI) occurs naturally, has a high water solubility, and is the main pollutant compound due to its carcinogenic, mutagenic and teratogenic effects on biological systems.7 At the moment, the World Health Organization (WHO) Guidelines give a provisional value for total chromium of 0.05 mg L−1.8 Hence, the efficient and economical treatment of Cr(VI)-containing water has become a challenging topic for both academia and industry.9 Up to now, the major technologies that exist for the removal of Cr(VI) contain membrane treatments,10 ion exchange,11 electro-chemical precipitation12 and adsorption.13–15 Amongst them, the efficiency achieved by membrane treatments is often quite low, there is a high cost associated with using ion-exchange resins, and electro-chemical precipitation usually generates a toxic sludge. In contrast, adsorption is an extremely efficient method and has attracted increasing interest towards chromium removal due to its significant advantages such as simplicity of design and operation, cost-effectiveness, high efficiency, recyclability of the adsorbent and no secondary pollution caused by the by-products. Within this context, fabrication of high-performance sorbents with practicality is urgently needed.16
Recently, some natural biopolymers, such as chitosan, alginate and guar gum, have been applied for the removal process of Cr(VI) ions from contaminated water.17–19 Among them, alginate is one of the most interesting natural polymers as it can be easily processed into various shapes, such as beads, threads and films. This polymer is a linear block copolymer consisting of uronic acid residues, namely, β-D-mannuronic and α-L-guluronic acid, linked by (1–4)-linkages.20 It must be said that the salts of alginic acid or alginate with monovalent ions (alkali metals and ammonium) are soluble, while those with divalent or polyvalent metal ions (except Mg2+) and the acid itself are insoluble.21 Furthermore, it also has other distinctive advantages, such as biodegradability, biocompatibility, non-toxicity and unique functional properties.22 Because of these features, alginate has gathered much interest for the adsorption of heavy metals,23–28 including Cr(VI) removal. For instance, Wu et al. have prepared mesoporous titania beads via an alginate biopolymer template method that were used as adsorbents to treat aqueous solutions containing Cr(VI), and the adsorption capacity was only 8.4 mg g−1.29 Also, Gopalakannan et al. synthesized Fe3O4@Alg–Ce magnetic composite beads by incorporating Fe3O4 particles onto an alginate biopolymer followed by cross-linking with Ce3+ ions. The magnetic alginate beads possessed a sorption capacity of 14.29 mg g−1 for Cr(VI) removal.30 Additionally, Ali Olad et al. have reported a method to synthesize an alginate–montmorillonite/polyaniline (Alg–MMT/PANI) nanocomposite by chemical oxidative polymerization of aniline in the presence of an alginate–montmorillonite nanocomposite dispersion for the removal of Cr(VI).31 As can be seen, the above mentioned works mainly focused on the application of alginate hydrogel beads for removal of pollutants from aqueous solutions. However, the utilization of alginate beads to remove Cr(VI) from effluents on-scale application has been hindered by operational limitations associated with its physical characteristics, such as a dense gel layer, poor mechanical strength and low rigidity.32 Particularly, the dense gel layer structure may cause higher mass transfer resistance between the adsorbent and adsorbate, resulting in restricted ion diffusion. Additionally, the use of alginate beads is further limited by its insufficient mechanical strength, usually leading to its rupture, increasing the resistance in continuous-flow contact vessels accordingly. More significantly, the adsorption capacity of Cr(VI) exhibited by hydrogel beads was commonly low owing to the repulsion of the same negatively charged carboxyl groups with negatively charged chromate in the aqueous solution. Therefore, overcoming these problems is greatly needed and would possibly be critical to its practical application towards Cr(VI) removal from aqueous solutions.
Unlike to alginate-derived hydrogel beads, carbon beads have attracted tremendous attention for its large surface area, mechanical stability, high porosity and tunable surface chemistry.33 More favorably, the carbon beads also maintain the prominent advantage of hydrogel beads, which is its monolithic characteristic. Obviously, monolithic materials at the macroscopic substance level can be handled much more conveniently for re-usage and practical application owing to its easy operational handling. Besides, employing a controllable carbonization treatment process could not only increase the porosity, leading to a decrease in ion-transport resistance, but could also considerably improve the mechanical stability of the hydrogel beads. As a consequence, the development of a facile method that can controllably convert alginate-based hydrogel beads to carbon beads with high porosity would open a new way to prepare a practical adsorbent for the removal of Cr(VI) from water on a large-scale application.
In this work, new carbon beads with a developed nano-network were successfully fabricated through combinational carbothermal reduction and simple acid treatment of Ca2+-gelled sodium alginate hydrogel beads. The material was characterized by FESEM, EDS, TEM, BET, XRD, FTIR, XPS and Raman spectroscopy. The beads were utilized for the removal of Cr(VI) from an aqueous system using a batch process, and the relevant parameters like pH, kinetics, isotherms and the effect of co-existing ions were investigated. The conceivable removal mechanism was also discussed. The removal of Cr(VI) from aqueous solution by the beads in a fixed-bed column was also investigated and the parameters on the column dynamics were discussed and optimized.
2. Experimental
2.1 Materials and chemicals
Sodium alginate and CaCl2 were purchased from Sinopharm Chemical Reagent Corporation (China). K2Cr2O7, CrCl3·6H2O, ethanol, HCl and NaOH were purchased from Tianjin Kermel Chemical Reagent Factory (China). All of the reagents were of analytical grade and were used as received without any further purification. Double-distilled water was used throughout this study. Various Cr(VI) solutions with different concentrations were prepared by dissolving K2Cr2O7 in double-distilled water.
2.2 Preparation of Ca–SA beads
Sodium alginate (8.0 g) was dissolved in 400 mL deionized water and stirred until evenly dispersed. The calcium alginate (CA) hydrogel beads with a mean diameter of 4 mm were obtained by drop-by-drop addition of prepared sodium alginate (2%, w/v) into CaCl2 (5%, w/v) aqueous solution under continual magnetic stirring. The obtained beads were immersed in the CaCl2 solution for another 6 h for complete gelation. The hydrogel beads were filtered and washed with deionized water to remove extra Cl− and Ca2+. In addition, the obtained hydrogel beads were washed with ethanol until they floated on the solution of the upper layer, and then dried in a 60 °C oven for 12 h.
2.3 Preparation of Alg-CB
The dried Ca–SA beads were pyrolyzed in a tube furnace at the designed temperature (400, 600 or 800 °C) in an N2 environment for 2 h. The resulting material (i.e. the precursor) was permitted to cool at room temperature under flowing nitrogen. Then, it was treated with 1 M HCl solution for 24 h to remove the Ca0 and other impurities at a maximum extent, and washed with distilled water until the supernatant was neutral, then dried in an oven at 60 °C. Finally, they were sealed to preserve freshness before use. The obtained materials were denoted as Alg-CB-X (X = 400, 600 or 800), corresponding to heating temperature. The preparation process of Alg-CB is shown in Scheme 1.
 |
| | Scheme 1 Schematic illustration of the Alg-CB fabrication process. | |
2.4 Materials characterization
X-ray diffraction (XRD) patterns were obtained with a Shimadzu XRD-6100 diffractometer with Cu-Kα radiation (λ = 1.540 Å) from 10° to 80° at an 8° min−1 scanning speed. The surface morphologies and the particle distribution of Alg-CB were determined by field emission scanning electron microscopy (FESEM, JSM-6460LV, JEOL, Japan) and transmission electron microcopy (TEM, JEM-2000EX electron microscope, JEOL, Japan), respectively. The surface elemental composition was obtained by X-Max50 energy dispersive X-ray spectroscopy (EDS, Oxford, UK) in the form of pressed pellets and selecting all the area in the field of the electron microscope. Fourier transform infrared (FTIR, Perkin-Elmer, USA) spectra in the 4000–500 cm−1 region were acquired using KBr pellets. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo Scientific ESCALAB250 spectrometer (Thermo VG, USA) equipped with an Al-Kα X-ray source (1486.6 eV). The specific surface area and pore diameter of the samples were performed by nitrogen adsorption–desorption experiments at 77 K (Quantachrome Autosorb NOVA2200e, USA). Raman spectrometry (RM2000, Renishaw, UK) was used to study the integrity of the sample. UV-Vis adsorption spectra were recorded on a MAPADA UV-1600PC spectrometer. Atomic absorption spectroscopy was employed by a Hitachi 180-80, Japan.
2.5 Chromium analysis
A colorimetric method, as described in the standard methods, was used to measure the concentrations of the different chromium species. The pink colored complex, formed from 1,5-diphenylcarbazide and Cr(VI) in acidic solution, was spectrophotometrically analyzed at 540 nm. Total Cr concentration was determined by Atomic Absorption Spectrometry (AAS). The concentration of trivalent chromium was calculated as the difference between total chromium and hexavalent chromium.
2.6 Removal experiments
2.6.1 Batch removal studies. The removal of Cr(VI) from aqueous systems onto Alg-CB was studied in a batch system. All experiments were performed in triplicate with appropriate blanks and benchmarks. In each removal experiment, 20 mL of Cr(VI) solution with the desired concentration was put into a 50 mL conical flask. The solution pH was measured by a pH meter and was adjusted to a predetermined value with 0.1 M HCl or 0.1 M NaOH. Then, 20 mg Alg-CB was added into the above solution. Subsequently, the mixture was stirred in a magnetic shaker to reach equilibrium (except for the kinetic experiments). The stirring process ensured the homogeneity of the system in the removal process. At the end of each experiment, the supernatant can be directly transferred with a pipette from the conical flask.The amount of total Cr, Cr(VI) + Cr(III), and removal at equilibrium, Qcrt (mg g−1), was calculated from the equation:
where
V (L) is the volume of the metal solution,
C0 (mg L
−1) is the initial concentration of total Cr in solution (equal to Cr(
VI)),
Ce (mg L
−1) is the equilibrium concentration of total Cr in solution, and
m (g) is the mass of sorbent (dry weight).
2.6.2 Column removal studies. Fixed-bed column operation was conducted in a glass column having a length of 12 cm and an internal diameter of 3.5 mm. A certain amount of degreasing cotton was kept at the bottom and top of the column to stabilize the adsorbents in the column. The designed concentration of Cr(VI) solutions was pumped into the column downward, using a peristaltic pump at a fixed flow rate, that was packed with 0.356 g Alg-CB at a bed height of 10 cm. The samples were collected at different time intervals from the effluent over eight hours and were analyzed.The fixed-bed removal capacity, qc (mg), is equal to the area under the plot from the integral of removed concentrations expressed as Cad (Cad = C0 − Ct) mg L−1 for a given time t (min) and was calculated as follows:
| |
 | (2) |
where
Q and
A are the effluent flow rate (mL min
−1) and the area under the breakthrough curve, respectively, and
t is the total flow time (min).
The equilibrium uptake, q, the weight of Cr(VI) removed per unit of dry adsorbent weight can be evaluated using:
where
X (g) is the mass of the total adsorbents in the column.
3. Results and discussion
3.1 Characterization of Alg-CB-X
The surface morphology of the precursor, HCl treated carbon beads (i.e. Alg-CB) and the beads after Cr removal (denoted as Alg-CB-Cr) were studied by FESEM (see Fig. S1†). From Fig. S1a and b,† it can be seen that the surface of the samples consisted of many nanometer particles and disordered mesostructures in large domains with fully open pore channels on the surfaces for the precursor and Alg-CB. Also, the distribution of Alg-CB surface pores was more than the precursor; meanwhile, the pore size of Alg-CB was larger and more unobstructed than that of the precursor, which was possibly attributed to the HCl treatment process from the precursor to Alg-CB. As a result, the pore structure would provide enough surface, benefiting the combination of the adsorption sites and Cr(VI) ions, and it could also prove Alg-CB possessed an open nano-network structure. In addition, as depicted in Fig. S1b and c,† by comparing the surface morphology of Alg-CB and Alg-CB-Cr, it can be found that no obvious changes of the bead surface appeared after removal of Cr(VI). This demonstrated that the surface of the adsorbent was not damaged by highly oxidizing Cr(VI) and it can be easily regenerated by means of desorption.
Also, EDS was conducted to investigate the surface elemental state of the precursor, Alg-CB and Alg-CB-Cr. From Fig. S2a and b,† it was clear that the carbon content increased from 33.33 to 96.93 (wt%), while the content of calcium and other impurities decreased dramatically after HCl treatment. Moreover, after the removal of Cr(VI) onto the Alg-CB, the adsorbent surface showed characteristic peaks of Cr (about at 0–1 keV) in the EDS (see Fig. S2c†), confirming that a Cr species had been deposited onto the surface of Alg-CB beads.
Afterwards, the microstructural characterization of Alg-CB-X (X = 400, 600 and 800) would be helpful in correlating the properties that may affect the removal of Cr(VI). Therefore, the morphology and microstructure of Alg-CB-X were further investigated by TEM. As seen in Fig. S3,† a large number of mesopores are uniformly distributed throughout the carbon framework. By contrast, the porosity of the ones that were not completely carbonized under low temperature were less developed, which would be unfavorable to the removal of Cr(VI) (Fig. S3a†). With increasing carbonizing temperature, the porosity was significantly developed; meanwhile, the carbon material became loosened, revealing that carbon beads with a developed nano-network had been prepared by employing an HCl treatment of carbonized Alg-CB (Fig. S3b and c†).
As a consequence, N2 absorption–desorption measurements were carried out to examine the surface area and the porous structures of the Alg-CB-X and Alg-CB-800-Cr. As depicted in Fig. S4a,† a hysteretic loop of the desorption branch over the middle of pressure, which can be ascribed to the capillary condensation at mesopores, was observed. It revealed that the BET surface areas of the four samples were 112.2, 410.5, 444.0 and 405.2 m2 g−1, respectively. Detailed data are summarized in Table S1.† Clearly, the specific surface areas of Alg-CB-X increased with increasing carbonization temperature. The increased surface areas may have been caused by a decrease in the size of the inter-particles and the expansion of gases such as H2O and CO2 formed through the interaction between carbon and calcium oxides in the material at higher temperature treatments under an N2 atmosphere. In addition, the surface area of Alg-CB-800-Cr was less than Alg-CB-800 and this may be because the pores were occupied by Cr(VI) ions. The pore size distribution is shown in Fig. S4b.† The four samples possessed a large number of mesopores, with the diameter located at 3.5–15.5 nm. The mesopores that were distributed throughout the carbon walls would provide a large internal surface, which was advantageous for mass transfer between the adsorbent and Cr(VI) ions.34
Furthermore, the crystalline structures of Alg-CB-X were characterized by XRD. As is seen in Fig. S5a,† a strong peak at 2θ = 23.5°, which was associated with the crystalline graphitic (200) plane, is presented while the weak peak at 2θ = 44° can be due to the crystalline graphitic (101) plane, indicating the presence of disordered graphitic carbon in the final sample.35 There is no other impurity peak, such as calcium, indicating the complete removal of metal residues. The specific nature can be further elucidated by Raman spectroscopy and Fig. S5b† presents the Raman spectra for the same carbon material. All samples showed a D peak at 1343 cm−1 and 1595 cm−1 for the G band. The D (defect) band (1343 cm−1) is due to the breathing mode of k-point phonons of A1g symmetry and the G (graphitic) band (1595 cm−1) was assigned to the E2g phonon of sp2 carbon atoms.36 Generally, the intensity ratio of the D-band to G-band (ID/IG) can be used to estimate the degree of carbon disorder;37 the high ID/IG ratio indicates the presence of disorder in the carbon matrix.36 The intensity ratios of ID/IG were 0.33, 0.49 and 0.71 for Alg-CB-400, Alg-CB-600 and Alg-CB-800, respectively. Comparatively, with the temperature increasing, the value of ID/IG became larger, implying that the carbon atoms of the Alg-CB-800 were more amorphous in nature, possibly leading to a more disordered bond at the atomic level. Surely, this would be beneficial to the removal of Cr(VI) on account of the edge defects of carbon materials, and they can be considered as active sites that can very easily interact with polar groups.38
3.2 Batch removal studies
3.2.1 Effect of pH on Cr(VI) removal. As is reported, Cr(VI) exists in the solution as various species that are entirely dependent on the pH of the solution and its concentration. Furthermore, from acidic pH = 1 to neutral pH = 7, the hydrogen chromate ion (HCrO4−) exists; whereas, above neutral pH, only chromate ions (CrO42−) exist in the solution.39 As a consequence, the dependence of Cr(VI) removal over Alg-CB-800 on the solution pH can be due to pH influences on the surface charge of the adsorbent. When the pH value is low, Alg-CB-800 static charges were present in a positively charged form. However, more and more negative charge formed on the surface of the adsorbent with increasing pH values. Considering the dominant species of Cr ion in solution was HCrO4− under relatively acidic conditions, accordingly, the chromate anion would have interacted strongly with the positively charged ions of the Alg-CB-800. Meanwhile, a low pH also accelerates the redox reactions since the protons take part in this reaction. As shown in Fig. 1, pH significantly affected Cr(VI) removal. Obviously, with increasing pH, the concentration of Cr(VI) in the aqueous solution increased while the concentration of Cr(III) decreased. Additionally, the concentration of total Cr at pH ∼ 2.0 was higher than the amount at pH ∼ 3.0 due to the formation of Cr(III), and the concentration of total Cr drastically increased when the pH increased from 3.0 to 10.0.
 |
| | Fig. 1 Effect of pH on the removal of Cr(VI) by Alg-CB-800 (material mass, 0.02 g; volume, 20 mL; initial Cr(VI) concentration, 25 mg L−1; contact time, 4 h and temperature, 293 ± 2 K). | |
3.2.2 Effect of contact time on Cr(VI) removal. For optimizing the maximum Cr(VI) removal capacity, the removal experiment was carried out by varying contact time between 10 and 7200 min with 25 mg L−1 as the initial Cr(VI) concentration and 0.02 g as the adsorbent mass at a solution pH of 3.0. The curves of Cr(VI) removal by Alg-CB-X were shown in Fig. 2. As seen from the plot, the concentrations of total Cr and Cr(VI) obviously declined with increasing contact time in 840 min. However, the concentration of Cr(III) keeps close to zero when contact time was increased from 10 to 840 min. The reason is that the adsorbed Cr(VI) remains bound to the functional groups that reduce Cr(VI) very slowly because of the lack of oxygen-containing functional groups, caused by the pyrolysis process, and the short adsorption period. Thus, the decreasing concentration of Cr(VI) was completely due to the removal of the Cr(VI) anion with the positively charged material surface. The concentration of Cr(III) increased 840 minutes later when the contact time increased, which was attributed to the reduced Cr(III) from the accumulation of small amounts of oxygen-containing functional groups. Furthermore, the adsorption equilibrium time of Alg-CB-800 was about 240 min and the removal capacity was kept constant after further increasing the contact time to 840 min. In contrast, the Alg-CB-600 and Alg-CB-400 all required about 360 min for equilibration. More significantly, the Alg-CB-800 possessed an enhanced removal capacity of 24.5 mg g−1 over both Alg-CB-600 and Alg-CB-400 samples, which possessed capacities of 23.9 and 10.8 mg g−1, respectively.
 |
| | Fig. 2 Effect of contact time on the removal of Cr(VI) by (a) Alg-CB-400, (b) Alg-CB-600, (c) Alg-CB-800 and (d) the experimental data for Cr(III) concentration in solution at different contact times (material mass, 0.02 g; initial Cr(VI) concentration, 25 mg L−1; volume, 20 mL; pH, 3.0 and temperature, 293± 2 K). | |
3.2.3 Effect of initial concentration on Cr(VI) removal. To start, 20 mL of Cr(VI) solutions with initial concentrations of 5–300 mg L−1 were prepared in a series of flasks to study the effect of initial Cr(VI) concentration on the removal process; and 0.2 g of Alg-CB-X was added into each flask. Equilibrium experiments were carried out at room temperature (20 °C) for 24 hours, the solution pH was adjusted to 3 and the results are shown in Fig. 3. It was found that the removal capacity of total Cr was strongly dependent on the initial metal ion concentration. Meanwhile, the equilibrium removal capacities increased with increasing initial Cr(VI) concentration. In addition, comparing the removal capacities of total Cr by Alg-CB-400, Alg-CB-600 and Alg-CB-800, the equilibrium removal capacities of Alg-CB-800 were much higher under each initial Cr(VI) concentration than that of other two samples. Consequently, the removal capacity of Alg-CB-400, Alg-CB-600 and Alg-CB-800 were 19.3, 37.1, and 50.4 mg g−1, respectively. This indicates that the heating temperature during the carbonization process was crucial toward the removal performance by the final sample, and this might be associated with the more developed nano-network of resulting beads. Additionally, the concentration of Cr(III) increased with increasing the initial concentration, and it was obviously higher with increased contact time for Cr(VI) removal.
 |
| | Fig. 3 Effect of initial concentration on the removal of Cr by (a) Alg-CB-400, (b) Alg-CB-600 and (c) Alg-CB-800 and (d) the experimental data for Cr(III) concentration in solution at different initial Cr(VI) concentrations (material mass, 0.02 g; volume, 20 mL; pH, 3.0; contact time, 24 h and temperature, 293± 2 K). | |
3.2.4 Effect of coexisting anions strength. Obviously, Cr(VI) ions exist in the form of anions in the aqueous phase, and the adsorption process of Cr(VI) occurs on the adsorbent surface through electrostatic interactions. Thus, the removal capacity of Cr(VI) would have been influenced by the ions existing in the solution. Therefore, competitive experiments were performed to study the effect of commonly present anions such as Cl−, NO3−, PO43−, and SO42− (C0 = 5, 10, 20 and 50 mmol L−1) on Cr(VI) uptake. As shown in Fig. 4, all four anions have an influence with differing levels, which is reflected by the following sequence of the removal capacity of Cr(VI): Cl− > NO3− > SO42− > PO43−. The Cl− and NO3− had less effect on the removal of Cr(VI). With increasing the concentration of Cl− and NO3−, the removal capacity decreased slightly. When the concentration of SO42− and PO43− increased from 5 to 50 mmol L−1, the removal capacity decreased dramatically; and the effect of PO43− was much larger than SO42−. The competition mechanism can explain this phenomenon.40 Cl− and NO3− are monovalent anions, and they may slightly compete with the Cr(VI) anion for positive charge adsorption sites on the Alg-CB-800 surface. However, the SO42− and the PO43− are multivalent anions that could compete with HCrO4−, Cr2O72− and CrO42− for more of the available sorption sites on the Alg-CB-800. The study of coexisting anion strength on Cr(VI) uptake demonstrated that the electrostatic force is one of the possible mechanisms for the removal of Cr(VI).
 |
| | Fig. 4 Effect of coexisting anion strength on the total Cr (material mass, 0.02 g; volume, 20 mL; initial Cr(VI) concentration, 25 mg L−1; contact time, 4 h and temperature, 293 ± 2 K). | |
3.3 Kinetic studies
In order to understand the kinetics of Cr(VI) adsorption by Alg-CB-X, pseudo first-order,41 pseudo second-order,42 Elovich43 and intraparticle diffusion44 kinetic models were fitted to the experimental data, which were measured in 600 min. The rate equations were expressed as follows:| | |
Qt = k2Qe2t/(1 + k2Qet)
| (5) |
| |
Qt = [ln(ab) + ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) t]/b t]/b
| (6) |
where Qe (mg g−1) and Qt (mg g−1) were the Cr(VI) removal amounts at the equilibrium state and time t (min), respectively; k1 (min−1) is the rate constant of the pseudo-first-order kinetic model; k2 (g mg−1 min−1) is the rate constant of the pseudo-second-order kinetic model; a (mg g−1 min−1) is the initial removal rate, and b (g mg−1) is related to the extent of surface coverage and the activation energy for the chemisorptions; Ki is the intraparticle constant and C is the intercept.
The kinetic parameters obtained are listed in Table 1 and fitted by the four kinetic models at a temperature of 293 K. Then, the curves were depicted in Fig. 5. It can be observed that the data can be fitted better with the pseudo-second-order model with a correlation coefficient more than 0.992, which indicated that the removal process was controlled by chemical adsorption.45 Also, due to the high porosity of the materials, surface diffusion (film diffusion) in addition to pore diffusion (intraparticle diffusion) may contribute to the overall Cr(VI) removal.46 In Fig. 5d, step 1, 2 and 3 are related to external surface diffusion, the pore diffusion within carbon beads and the equilibrium stage, respectively. The correlation coefficient of step 1 and step 2 were both high, indicating that boundary layer diffusion and intraparticle diffusion occurred in the removal process. Moreover, the slope of step 1 was higher than step 2, revealing the external diffusion stage was faster than the intraparticle diffusion stage. In addition, the Qt versus t0.5 plots for both step 1 and step 2 did not pass through the origin and a positive intercept was observed. This implies that some other removal processes, such as diffusion of the liquid boundary layer around the material toward the particle surface, might also be involved in the Cr(VI) removal.47
Table 1 Kinetic parameters of Cr(VI) removal using Alg-CB-X
| Kinetic |
Parameters |
Alg-CB-400 |
Alg-CB-600 |
Alg-CB-800 |
| Pseudo-first-order |
Qe,exp (mg g−1) |
10.6 ± 0.2 |
23.7 ± 0.1 |
24.5 ± 0.2 |
| k1 (min−1) |
0.012 ± 0.001 |
0.027 ± 0.002 |
0.036 ± 0.002 |
| Qe,cal (mg g−1) |
10.3 ± 0.2 |
23.3 ± 0.3 |
24.3 ± 0.3 |
| R2 |
0.9786 |
0.9427 |
0.9516 |
| Pseudo-second-order |
k2 (g mg−1 min−1) |
0.001 ± 0.000 |
0.002 ± 0.000 |
0.002 ± 0.001 |
| Qe,cal (mg g−1) |
12.1 ± 0.2 |
24.9 ± 0.1 |
25.8 ± 0.2 |
| R2 |
0.9848 |
0.9947 |
0.9920 |
| Intraparticle-diffusion |
Ki1 |
0.822 ± 0.015 |
2.824 ± 0.034 |
2.180 ± 0.011 |
| C1 |
−0.481 ± 0.021 |
−0.319 ± 0.033 |
1.468 ± 0.002 |
| R12 |
0.9884 |
0.9426 |
0.9935 |
| Ki2 |
0.450 ± 0.004 |
0.633 ± 0.083 |
0.645 ± 0.009 |
| C2 |
2.766 ± 0.118 |
15.06 ± 0.35 |
12.95 ± 0.19 |
| R22 |
0.9988 |
0.9658 |
0.9792 |
| Ki3 |
0.007 ± 0.001 |
0.043 ± 0.003 |
0.071 ± 0.001 |
| C3 |
10.63 ± 0.18 |
23.80 ± 0.25 |
22.30 ± 0.12 |
| R32 |
0.8677 |
0.7761 |
0.2125 |
| Elovich equation |
a |
0.559 ± 0.130 |
6.259 ± 2.332 |
7.249 ± 3.410 |
| b |
0.420 ± 0.032 |
0.279 ± 0.020 |
0.269 ± 0.026 |
| R2 |
0.9339 |
0.9400 |
0.9017 |
 |
| | Fig. 5 The pseudo-first-order (a), pseudo-second-order (b), Elovich (c) and intraparticle diffusion (d) kinetic models of Cr(VI) removal on Alg-CB-X (X represents 400, 600 and 800). | |
3.4 Adsorption equilibrium isotherm studies
This section mainly studied the adsorption process because the reduction process could not use the standard model for optimization. Equilibrium isotherms are very important in order to design adsorption processes; they also provide the adsorption capacity of the materials under the studied conditions. Because the removal mechanism processes consisted of adsorption and reduction, the equilibrium of the system can be divided in adsorption equilibrium and reduction equilibrium. According to the finished experiments, the adsorption process was in equilibrium at 24 h while the reduction process was in equilibrium at 80 hours. Thus, the removal process was in equilibrium at 80 hours. Meanwhile, the experimental data and the proposed mechanism both support that Cr(III) concentration in solution is negligible before the contact time reaches to 24 h, which means the adsorption process plays a major role in the early removal stage. Therefore, the adsorption equilibrium experiments mainly studied the first 24 hours since Cr(VI) could be taken away from the solution rather than be transformed into Cr(III) by the material, which would help us to optimize the data to evaluate the practical application of the material. Although there are many isotherm models, the Langmuir, Freundlich and Temkin models are the most frequently used equations in the literature that express the nonlinear relationship between metal ion on the adsorbent and metal ion in the solution. These three-parameter models are simple and give a good description of the experimental behavior in a large range of operating conditions.48 Langmuir, Freundlich and Temkin isotherm models were examined to describe the removal equilibrium of total Cr using three different adsorbents (Alg-CB-400, Alg-CB-600 and Alg-CB-800) at a certain pH and temperature.
The Langmuir theory was the first adsorption isotherm model introduced on the basis of a kinetic viewpoint in 1918.49 The Langmuir model is expressed by the following equation:
| | |
Qe = QmKLCe/(1 + KLCe)
| (8) |
where
Qe is the amount of metal ions removed per unit mass of adsorbent (mg g
−1),
Ce is the equilibrium concentration of the solute in the bulk solution (mg L
−1),
Qm and
KL are Langmuir constants, respectively the removal capacity (mg g
−1) and the energy of removal (L mg
−1).
The Langmuir isotherm can be expressed in terms of a dimensionless separation factor, RL, which describes the type of isotherm;
where
C0 is the initial concentration of Cr(
VI). The magnitude of
RL determines the feasibility of the removal process. If
RL > 1, the removal is unfavorable; if
RL = 1, the removal is linear; if
RL < 1, the removal is favorable and if
RL = 0, the removal is irreversible.
The second model applied to the adsorption isotherm was the Freundlich model, which was one of the earliest known empirical equations based on a heterogeneous surface and adsorption heat.50 It is expressed by the following equation:
where
KF and
n are indicators of removal capacity and removal intensity, respectively. For a good adsorbent, the value of 1/
n is less than 1.
The third model used for the adsorption isotherm was the Temkin model, which is a proper model for chemical adsorption based on strong electrostatic interactions between positive and negative charges.51 It is expressed by the following equation:
| | |
Qe = Bln(ACe)
| (11) |
where
A (L mg
−1) and
B (mg g
−1) are Temkin isotherm constants.
The results of fitting these models are shown in Fig. 6 and the fitting parameters are listed in Table 2. The value of the correlation coefficient (R2) ranged from 82.85% to 95.19% and was higher for the Langmuir isotherm than for the Freundlich isotherm, which ranged from 61.51% to 79.30%. Thus, the removal isotherm of Cr(VI) for the three samples exhibited Langmuir behavior, indicating that the uptake occurred on surface by monolayer adsorption and can be described in terms of chemisorption, that is, the formation of electrostatic attraction between adsorbent and adsorbate. Since the value of 1/n was less than 1, it revealed a favorable adsorption process. While the Temkin models fitted reasonably, its correlation coefficient (R2) ranged from 80.61% to 93.39%, suggesting that there was some electrostatic interaction in the removal process.
 |
| | Fig. 6 Effect of initial concentration on the removal of total Cr by Alg-CB-X (a) (material mass, 0.02 g; volume, 20 mL; pH, 3.0; contact time, 24 h and temperature, 293± 2 K) and Langmuir, Freundlich and Temkin isotherm models of the total Cr removal using Alg-CB-400 (b), Alg-CB-600 (c) and Alg-CB-800 (d). | |
Table 2 Isotherm parameters of total Cr removal on Alg-CB-X at 293 K
| Isotherm models |
Parameters |
Alg-CB-400 |
Alg-CB-600 |
Alg-CB-800 |
| Langmuir |
Qm (mg g−1) |
23.1 ± 1.9 |
40.5 ± 3.7 |
56.2 ± 8.3 |
| KL (L mg−1) |
0.191 ± 0.067 |
0.164 ± 0.039 |
0.048 ± 0.010 |
| R2 |
0.9519 |
0.9264 |
0.8285 |
| RL |
0.0344–0.6814 |
0.0025–0.1328 |
0.0007–0.0394 |
| Freundlich |
KF (mg g−1) |
8.986 ± 3.046 |
6.055 ± 1.574 |
2.371 ± 0.809 |
| n |
2.817 ± 0.465 |
2.567 ± 0.418 |
2.396 ± 0.436 |
| R2 |
0.7930 |
0.7843 |
0.6151 |
| Temkin |
A (L mg−1) |
2.196 ± 0.598 |
2.076 ± 0.343 |
0.550 ± 0.093 |
| B (mg g−1) |
10.1 ± 1.6 |
7.4 ± 0.7 |
4.6 ± 0.4 |
| R2 |
0.9339 |
0.9202 |
0.8061 |
3.5 Column removal studies
The effect of flow rate was investigated by varying the flow rate (2.0, 4.0, 6.0 mL min−1) at a constant bed height (10.0 cm). The breakthrough curve of Ct/C0 versus time with varying flow rate is depicted in Fig. 7. The breakthrough time (tb) shows the time at which the outlet metal concentration reached the maximum allowable value (5% of inlet concentration) and the exhaustion time (te) represents the time at which the outlet heavy metal concentration exceeded 95% of the inlet metal concentration.52 The results indicated that a decrease in flow rate at a constant bed height increased the breakthrough time (tb). The bed exhaustion time (te) decreased with an increase in flow rate from 600 min (2.0 mL min−1) to 480 min (4.0 mL min−1) to 240 min (6.0 mL min−1). At higher flow rates, the residence time of the solution in the column is less; and hence, Cr ions have less contact with the material to diffuse into its pores. When the residence time of the solution was less in the column, equilibrium was not reached; thus, the Cr solution leaves the column before attaining equilibrium.53 On the other hand, lower flow rates increase the residence time and allows Cr ions to diffuse and reach the active sites of the material. The Cr ions adsorption capacity was 45.2, 28.4 and 17.0 mg g−1 with increasing flow rate for 2.0, 4.0 and 6.0 mL min−1, respectively. These results indicate that a slower flow rate and longer contact time is required for Cr removal in the Alg-CB-800 column. In addition, column experiments also revealed that continuous flow conformation is not suitable for Cr(VI) removal under the tested conditions (inlet concentration, 50 mg L−1; flow rates, 2, 4 and 6 mL min−1). Thus, a lower solution concentration and slower flow rate were sensible options. Alternatively, the column specifications could be increased while the solution concentration and flow rates remain unchanged.
 |
| | Fig. 7 The breakthrough curves of total Cr removal by Alg-CB-800 fixed-bed columns predicted by the Thomas and Adams–Bohart model at different flow rates (material mass, 0.356 g; initial Cr(VI) concentration, 50 mg L−1; pH, 3.0 and temperature, 293 ± 2 K). | |
The design and optimization of the breakthrough curve for a full-scale column removal system requires a simple modeling approach that can provide accurate scale-up column data. In the present study, the Thomas and Adams–Bohart model were employed to analyze the breakthrough curves for single metal systems. The Thomas model is one of the more widely applied models for continuous flow systems,54 which can be represented as:
| | |
ln(Ct/C0 − 1) = kTHq0w/Q − kThq0t
| (12) |
where
C0 (mg L
−1) is the influent metal ion concentration,
Ct (mg L
−1) is the effluent concentration at time
t.
kTh is the Thomas rate constant (L min
−1 g
−1) and
q0 is the maximum uptake capacity (mg g
−1).
Q (mL min
−1) is the flow rate and
w (g) is the sorbent mass. The model parameters,
kTh and
q0, can be estimated by non-linear fitting to the experimental data of the breakthrough curves.
The other model applied for correlating the breakthrough curve was the Adams–Bohart model,55 which is based on the surface reaction theory established in a fundamental equation. It was applied to experimental data to describe the initial part of the breakthrough curve, which was focused on the estimation of characteristic parameters such as maximum removal capacity (N0) and the kinetic constant (KAB). The expression is as follows:
| | |
ln(Ct/C0) = KABC0t − KABN0Z/F
| (13) |
where
KAB (mL min
−1 mg
−1) is the kinetic constant,
F (cm min
−1) is the linear velocity calculated by dividing the flow rate by the column section area,
Z (cm) is the bed depth of the column and
N0 (mg mL
−1) is the saturation concentration.
From Table 3, it is observed that, as the flow rate increases, the values of kTh and N0 increased, which represents that the mass transfer resistance present on the surface of the Alg-CB-800 could be significantly decreased. Additionally, the obtained non-linear regression correlation coefficients (R2) from the Adams–Bohart model were much less than those from the Thomas model. Thus, compared with the Adams–Bohart model, the Thomas model was a more suitable kinetic model to describe the total Cr uptake onto Alg-CB in a fixed-bed column.
Table 3 Parameters of Thomas and Adams–Bohart models analyzed for total Cr removal by Alg-CB-800 in a fixed-bed column
| Flow rate (mL min−1) |
tb (min) |
te (min) |
q (mg g−1) |
Thomas model constants |
Adams–Bohart model constants |
| kTh (L mg−1 min−1) |
q0 (mg g−1) |
R2 |
KAB (mL min−1 mg−1) |
N0 (mg mL−1) |
R2 |
| 2 |
15 |
480 |
45.2 ± 0.4 |
0.00031 ± 0.00002 |
41.888 ± 1.352 |
0.9370 |
0.074 ± 0.007 |
17.716 ± 0.115 |
0.7032 |
| 4 |
10 |
360 |
28.4 ± 0.4 |
0.00042 ± 0.00001 |
39.702 ± 0.983 |
0.8449 |
0.038 ± 0.004 |
24.303 ± 0.064 |
0.6449 |
| 6 |
5 |
240 |
17.0 ± 1.1 |
0.00050 ± 0.00002 |
33.473 ± 2.109 |
0.9581 |
0.016 ± 0.002 |
29.932 ± 0.040 |
0.5226 |
3.6 Removal mechanisms
In order to determine the main mechanism of Cr(VI) removal, XPS was used to detect the slight change of surface chemistry. As illustrated in Fig. 8a, the photoelectron lines at a binding energy of about 580, 532 and 285 eV were attributed to the Cr 2p, O 1s and C 1s, respectively. By comparing the curves before and after removal, high-resolution XPS spectra were collected from the Cr 2p core regions of the Cr-laden carbon beads as well as standard Cr(III) and Cr(VI) compounds. As shown in Fig. 8b, significant bands of a standard Cr(III) compound, CrCl3·6H2O, appeared at binding energies of 577–579 and 586–588 eV; the former corresponds to the Cr 2p3/2 orbital and the latter to the Cr 2p1/2 orbital. Meanwhile, those of a standard Cr(VI) compound, K2Cr2O7, appeared at binding energies of 579–581 and 588–590 eV, respectively. After interaction with Cr(VI), there was significant contribution of the chromium bound onto the carbon beads. Obviously, it can be seen that the Cr 2p1/2 and Cr 2p3/2 were located at 578 and 588 eV, respectively. Each of them could be further fitted into two peaks, the ones at 588.7 and 587.1 eV were assigned to the Cr 2p1/2 of Cr(VI) and Cr(III), while those at 579 and 577.5 eV were assigned to the Cr 2p3/2 of Cr(VI) and Cr(III), indicating that Cr(VI) and Cr(III) species coexisted on the surface of the sample. However, the reason why Cr(III) cations could coexist on the surface of the material is that the samples were measured by the XPS spectra were directly dried after the removal process. Consequently, some solvent remained in the carbon beads (water) that was evaporated, and the solutes (Cr(III) ions) were left on the material surface. Nonetheless, it was still demonstrated that the Cr(VI) could be reduced to Cr(III). Fig. 8c and d displays the O 1s spectra of Alg-CB-800 before and after Cr(VI) removal. The peaks at 533.38, 532.4 and 531.48 eV are ascribed to O–H, O–C and O–Ca, respectively. Comparing before and after removal, a new peak appeared at 532.09 eV, which was attributed to O–Cr by the adsorption of Cr(VI) anions onto the Alg-CB-800 surface. The intensity of O–H, O–C and O–Ca has changed to a certain extent because of the existence of O–Cr. The presence of oxygen from before and after removal explained that functional groups could be recovered in the reduction of Cr(VI) to Cr(III). After the removal process, the C–H and C–OH function groups could have reduced the Cr(VI) to Cr(III); meanwhile, it would be oxidized to C–OH and –COOH, respectively. Afterward, the –COOH would be transformed into –C–H or –C–OH for its easy decomposition. Finally, the formed Cr(III) was released into the aqueous phase due to electronic repulsion between the positively-charged groups and the Cr(III).56
 |
| | Fig. 8 XPS spectra of (a) the survey before and after removal (mass, 0.4 g; volume, 200 mL; contact time, 24 h; initial Cr(VI) concentration, 400 mg L−1; pH, 3.0 and temperature, 293 ± 2 K), (b) the Cr 2p core regions of the Cr-laden carbon beads as well as standard Cr(III) and Cr(VI), the O 1s pattern of (c) before and (d) after removal. | |
Moreover, FT-IR spectra of the precursor, Alg-CB-800, and Alg-CB-800-Cr are depicted in Fig. S6.† As is seen in the plot before and after HCl treatment (the precursor and Alg-CB-800), the two spectra showed analogous peaks except for the two bands at around 563 and 605 cm−1, which can be attributed to the Ca–O component in the precursor. The two bands at around 3440 and 1044 cm−1 can be assigned to O–H and C–O stretching vibrations, respectively, indicating the presence of a certain amount hydroxyl groups. While contrasting before and after removal spectra, the band intensity at 3440 cm−1 drastically decreases, indicting the hydroxyl groups have been consumed during the removal process. The band at 1549 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration. A sharp peak at 2923 cm−1 and a small peak at 2852 cm−1 are attributed to the C–H stretching and bending vibrations. Considering the fact that the solution pH could make an impact on the reduction ability of C–H bonds of the samples, the reduction ability of C–H groups would be promoted when the system is weakly acidic. Therefore, it was possible that oxidation also participated in the removal process, leading to the reduction of Cr(VI) to Cr(III). However, no COOH groups that could decompose easily under high temperature were observed on the Cr(VI)-laden Alg-CB-800. The redox reaction and the generation and decomposition of COOH groups may be explained by the following equations:57
C stretching vibration. A sharp peak at 2923 cm−1 and a small peak at 2852 cm−1 are attributed to the C–H stretching and bending vibrations. Considering the fact that the solution pH could make an impact on the reduction ability of C–H bonds of the samples, the reduction ability of C–H groups would be promoted when the system is weakly acidic. Therefore, it was possible that oxidation also participated in the removal process, leading to the reduction of Cr(VI) to Cr(III). However, no COOH groups that could decompose easily under high temperature were observed on the Cr(VI)-laden Alg-CB-800. The redox reaction and the generation and decomposition of COOH groups may be explained by the following equations:57
| –C–H + Cr(VI) + H2O → –C–OH + Cr(III) + H+ |
| –C–OH + Cr(VI) + H2O → –CO + Cr(III) + H+ |
| –CO + Cr(VI) + H2O → –COOH + Cr(III) + H+ |
| –C–COOH + Cr(VI) + H2O → –C–H + Cr(III) + H+ + CO2↑ |
which was consistent with the former XPS analysis results. Therefore, the surface structure of Alg-CB-800 could remain during the removal process and the amount of reducing groups on the material did not decrease after regeneration, which would be beneficial to its reuse.
Based on the kinetic, isotherm and removal mechanism analysis, the whole process of Cr(VI) removal should consist of the following steps: firstly, the external mass transfer of Cr(VI) anions occurs from the bulk solution to the material surface through electrostatic attraction between the protonated oxygen-containing function groups and the negative HCrO4−. Then, the Cr(VI) ions diffuse into the internal pores of carbon beads and part of them are reduced by C–H or C–OH groups. It is emphasized that a portion of the adsorbed Cr(VI) remains bound to the functional groups that reduce Cr(VI) very slowly because of the lack of oxygen-containing functional groups, caused by the pyrolysis process, and the short removal period. With increasing experimental time and initial concentration, reduced Cr(III) gradually forms. Finally, Cr(VI) is deposited onto the carbon surface, and the formed Cr(III) is released into the aqueous phase due to electronic repulsion between the positively-charged groups and the Cr(III). The possible adsorption mechanism is depicted in Scheme 2.
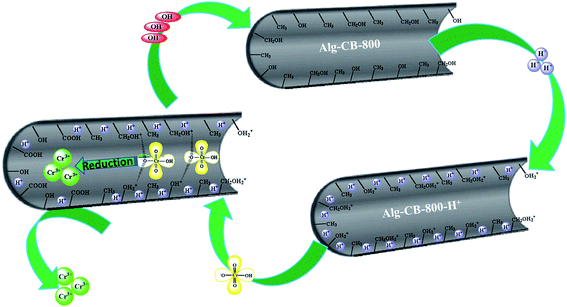 |
| | Scheme 2 The possible mechanism of Cr(VI) removal by Alg-CB. | |
3.7 Desorption and reusability study
To make the uptake media cost effective for Cr(VI) removal from aqueous systems, it is important that the samples should be reused for repeated cycles. For repeated use of an adsorbent, removed metal ions should be easily desorbed under suitable conditions. Desorption of the adsorbed Cr ions from the sample was studied in a batch experimental system and it is depicted in Fig. S7.† Desorption experiments put into evidence that, after 2 h contact with 20 mL 1 M NaOH solutions, it's sorption capacity could be nearly recovered. The recycling test of NaOH solution-treated Alg-CB-800 showed that the adsorption–desorption process was reversible. Three cycles of adsorption–desorption experiments were conducted to examine the capacity of the sample to retain Cr removal capacity. The removal capacity of the material decreased less than 20% during a three adsorption–desorption test cycle. The decline of the performance may be ascribed to the fact that the reduction property of the material to Cr(VI) was diminished after the first removal. In the subsequent cycles, the removal capacity was basically kept constant. Desorption and recyclability studies indicated that Alg-CB-800 could be repeatedly used as an efficient adsorbent in the process of removing Cr ions.
3.8 Comparison of adsorption capacity with literature data
The Alg-CB-800 adsorbent was compared in terms of removal capacity with other similar alginate-based and carbon materials. From the results (Table S2†) it is evident that the removal capacity of the proposed adsorbent compares favorably with other alginate-based adsorbents, and its removal capacity has taken great strides.
4. Conclusion
Novel alginate-derived carbon beads with a developed nano-network were successfully synthesized, through a facile carbothermal reduction and simple acid treatment method, for the easy separation and removal of Cr(VI) ions from aqueous solutions. The batch removal experiments showed that the removal of Cr(VI) was dependent on solution pH, contact time, initial Cr concentration and the existence of coexisting anions. The mechanistic investigation revealed that Cr(VI) uptake onto Alg-CB-X involved electrostatic interactions and slightly involved redox reactions. However, it is emphasized that the reduction process of Cr(VI) to Cr(III) was very slow and slight because of the lack of oxygen-containing functional groups, caused by pyrolysis process, and the short adsorption period. The removal kinetics followed a pseudo-second-order kinetic equation, and the equilibrium data could be fitted by the Langmuir isotherm. The reusability test showed that the material can be reused easily. The column removal studies showed the Cr(VI) uptake capacity decreased by increasing the flowing rate, and slightly less than that of batch studies. The Thomas and Adams–Bohart models predicted the removal capacity of Cr(VI) by the adsorbents in a fixed bed column. It was demonstrated that the material was an effective adsorbent for Cr(VI) removal with quick separation; and, most importantly, this kind of Alg-CB could be handled much more conveniently for reuse and scale-up for the practical applications, compared to conventional powdery adsorbents, owing to its easily operability and separation.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21546008, 21676039), the Program for Liaoning Innovative Research Team in University (LT2013012) and the Program for Liaoning Excellent Talents in University (LJQ2014056) is highly appreciated.
References
- A. Nematollahzadeh, S. Seraj and B. Mirzayi, Chem. Eng. J., 2015, 277, 21–29 CrossRef CAS.
- Z. Q. Ren, D. L. Kong, K. Y. Wang and W. D. Zhang, J. Mater. Chem. A, 2014, 2, 17952–17961 CAS.
- B. Qiu, H. B. Gu, X. R. Yan, J. Guo, Y. R. Wang, D. Z. Sun, Q. Wang, M. Khan, X. Zhang, B. L. Weeks, D. P. Young, Z. H. Guo and S. Y. Wei, J. Mater. Chem. A, 2014, 2, 17454–17462 CAS.
- S. Wang, W. Yang and G. Chen, RSC Adv., 2015, 5, 65982–65990 RSC.
- Y. Chen and G. Gu, Bioresour. Technol., 2005, 96, 1713–1721 CrossRef CAS PubMed.
- S. M. Doke and G. D. Yadav, Chem. Eng. J., 2014, 255, 483–491 CrossRef CAS.
- Z. Jiang, Y. Liu, G. Zeng, W. Xu, B. Zheng, X. Tan and S. Wang, RSC Adv., 2015, 5, 25389–25397 RSC.
- I. Polowczyk, B. F. Urbano, B. L. Rivas, M. Bryjak and N. Kabay, Chem. Eng. J., 2016, 284, 395–404 CrossRef CAS.
- Y. Du, Z. Tao, J. Guan, Z. Sun, W. Zeng, P. Wen, K. Ni, J. Ye, S. Yang, P. Du and Y. Zhu, RSC Adv., 2015, 5, 81438–81444 RSC.
- R. K. Goyal, N. S. Jayakumar and M. A. Hashim, J. Hazard. Mater., 2011, 195, 383–390 CrossRef CAS PubMed.
- S. Rengaraj, C. K. Joo, Y. Kim and J. Yi, J. Hazard. Mater., 2003, 102, 257–275 CrossRef CAS PubMed.
- N. Kongsricharoem and C. Polprasert, Water Sci. Technol., 1996, 34, 109–116 CrossRef.
- Y. Zhao, J. R. Peralta-Videa, J. R. Lopez-Moreno, M. Ren, G. Saupeand and J. L. Gardea-Torresdey, Environ. Sci. Technol., 2011, 45, 1082–1087 CrossRef CAS PubMed.
- D. Zhang, S. Y. Wei, C. Kaila, X. Su, J. Wu, A. B. Karki, D. P. Young and Z. H. Guo, Nanoscale, 2010, 2, 917–919 RSC.
- M. A. Barakat, Arabian J. Chem., 2011, 4, 361–377 CrossRef CAS.
- S. M. Lee, C. Laldawngliana and D. Tiwari, Chem. Eng. J., 2012, 195–196, 103–111 CrossRef CAS.
- F. Ferreroa, C. Tonettib and M. Periolatto, Carbohydr. Polym., 2014, 110, 367–373 CrossRef PubMed.
- D. Park, S. R. Lim, Y. S. Yun and J. M. Park, Bioresour. Technol., 2008, 99, 8810–8818 CrossRef CAS PubMed.
- E. S. Abdel-Halim and S. S. Al-Deyab, Carbohydr. Polym., 2011, 86, 1306–1312 CrossRef CAS.
- A. Konwar, A. Gogoi and D. Chowdhury, RSC Adv., 2015, 5, 81573–81582 RSC.
- C. Gok and S. Aytas, J. Hazard. Mater., 2009, 168, 369–375 CrossRef CAS PubMed.
- E. S. Abdel-Halim and S. S. Al-Deyab, Carbohydr. Polym., 2011, 84, 454–458 CrossRef CAS.
- Y. Kuang, J. H. Du, R. B. Zhou, Z. L. Chen, M. Megharaj and R. Naidu, J. Colloid Interface Sci., 2015, 447, 85–91 CrossRef CAS PubMed.
- B. An, H. Lee, S. Lee, S. H. Lee and J. W. Choi, J. Hazard. Mater., 2015, 298, 11–18 CrossRef CAS PubMed.
- A. Bée, D. Talbot, S. Abramson and V. Dupuis, J. Colloid Interface Sci., 2011, 362, 486–492 CrossRef PubMed.
- X. L. Li, Y. X. Qi, Y. F. Li, Y. Zhang, X. H. He and Y. Wang, Bioresour. Technol., 2013, 142, 611–619 CrossRef CAS PubMed.
- S. B. Hammouda, N. Adhoum and L. Monser, J. Hazard. Mater., 2015, 294, 128–136 CrossRef CAS PubMed.
- A. Idris, N. Suriani Mohd Ismail, N. Hassan, E. Misran and A. F. Ngomsik, J. Ind. Eng. Chem., 2012, 18, 1582–1589 CrossRef CAS.
- N. Wu, H. H. Wei and L. Zhang, Environ. Sci. Technol., 2012, 46, 419–425 CrossRef CAS PubMed.
- V. Gopalakannan and N. Viswanathan, Int. J. Biol. Macromol., 2015, 72, 862–867 CrossRef CAS PubMed.
- A. Olad and F. F. Azhar, Desalin. Water Treat., 2014, 52, 2548–2559 CrossRef CAS.
- H. Lee, D. Kim, J. Kim, M. K. Ji, Y. S. Han, Y. T. Park, H. S. Yun and J. Choi, J. Hazard. Mater., 2015, 292, 146–154 CrossRef CAS PubMed.
- W. Teng, Z. X. Wu, J. W. Fan, W. X. Zhang and D. Zhao, J. Mater. Chem. A, 2015, 3, 19168–19176 CAS.
- Y. Li, Q. L. Liu, D. M. Kang, J. J. Gu, W. Zhang and D. Zhang, J. Mater. Chem. A, 2015, 3, 21016–21022 CAS.
- X. He, X. Y. Peng, Y. X. Zhu, C. Lai, C. Ducatia and R. V. Kumar, Green Chem., 2015, 17, 4637–4646 RSC.
- M. Biswal, A. Banerjee, M. Deo and S. Ogale, Energy Environ. Sci. Technol., 2013, 6, 1249–1259 RSC.
- D. H. Li, C. X. Lv, L. Liu, Y. Z. Xia, X. L. She, S. J. Guo and D. Yang, ACS Cent. Sci., 2015, 1, 261–269 CrossRef CAS PubMed.
- I. Y. Jeon, Y. R. Shin, G. J. Sohn, H. J. Choi, S. Y. Bae, J. Mahmood, S. M. Jung, J. M. Seo, M. J. Kim, D. W. Chang, L. Dai and J. B. Baek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5588–5593 CrossRef CAS PubMed.
- M. K. Dinker and P. S. Kulkarni, J. Chem. Eng., 2015, 60, 2521–2540 CAS.
- G. Yang, L. Tang, Y. Cai, G. Zeng, P. Guo, G. Chen, Y. Zhou, J. Tang, J. Chen and W. Xiong, RSC Adv., 2014, 4, 58362–58371 RSC.
- S. Kabiri, D. N. H. Tran, S. Azari and D. Losic, ACS Appl. Mater. Interfaces, 2015, 7, 11815–11823 CAS.
- Y. S. Ho and G. Makay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- Z. M. Lei, S. R. Zhai, J. L. Lv, Y. Fan, Q. D. An and Z. Y. Xiao, RSC Adv., 2015, 5, 77932–77941 RSC.
- J. H. Zhu, S. Y. Wei, H. B. Gu, S. B. Rapole, Q. Wang, Z. P. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS PubMed.
- B. Qiu, J. Guo, X. Zhang, D. Z. Sun, H. B. Gu, Q. Wang, H. W. Wang, X. F. Wang, X. Zhang, B. L. Weeks, Z. H. Guo and S. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 19816–19824 CAS.
- S. Gupta and B. V. Babu, Chem. Eng. J., 2009, 150, 352–365 CrossRef CAS.
- Y. X. Zhao, W. F. Qi, G. Y. Chen, M. Jia and Z. Zhang, J. Taiwan Inst. Chem. Eng., 2015, 50, 190–197 CrossRef CAS.
- D. Duranǒglua, A. W. Trochimczuk and U. Beker, Chem. Eng. J., 2012, 187, 193–202 CrossRef.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. Freundlich, J. Phys. Chem., 1906, 57, 385–470 CAS.
- Y. Gao, Y. Li, L. Zhang, H. Huang, J. J. Hu, S. M. Shah and X. Su, J. Colloid Interface Sci., 2012, 368, 540–546 CrossRef CAS PubMed.
- R. Lakshmipathy and N. C. Sarada, Environ. Sci.: Water Res. Technol., 2015, 1, 244–250 CAS.
- T. Mayer-Gall, K. Opwis and J. S. Gutmann, J. Mater. Chem. A, 2015, 3, 386–394 CAS.
- H. C. Thomas, J. Am. Chem. Soc., 1944, 66, 1664–1666 CrossRef CAS.
- G. S. Bohart and E. Q. Adams, J. Am. Chem. Soc., 1920, 42, 523–544 CrossRef.
- D. Park, S. R. Lim, Y. S. Yun and J. M. Park, Chemosphere, 2007, 70, 298–305 CrossRef CAS PubMed.
- L. Z. Zhuang, Q. H. Li, J. S. Chen, B. B. Ma and S. Chen, Chem. Eng. J., 2014, 253, 24–33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Include EDS, effect of pH on the adsorption of Cr(VI), effect of contact time on the adsorption of Cr(VI), effect of initial concentration on the adsorption of Cr(VI), breakthrough curves, effect of coexisting anions strength on the Cr(VI) adsorption, FT-IR results, the reusability of Alg-CB-800 adsorbed Cr(VI) and BET parameters. See DOI: 10.1039/c6ra14070j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 










