
Catecholase activity, DNA binding and cytotoxicity studies of a Cu(II) complex of a pyridoxal schiff base: synthesis, X-ray crystal structure, spectroscopic, electrochemical and theoretical studies†
Piyali Adaka,
Bipinbihari Ghosha,
Antonio Bauzáb,
Antonio Frontera *b,
Alexander J. Blakec,
Montserrat Corbellad,
Chitrangada Das Mukhopadhyaye and
Shyamal Kumar Chattopadhyay*a
*b,
Alexander J. Blakec,
Montserrat Corbellad,
Chitrangada Das Mukhopadhyaye and
Shyamal Kumar Chattopadhyay*a
aDepartment of Chemistry, Indian Institute of Engineering Science and Technology, Shibpur, Howrah 711 103, India. E-mail: shch20@hotmail.com; Fax: +91 033 2668 2916
bDepartment of Chemistry, University of the Balearic Islands, Carretera de Valldemossa km 7.5, 07122 Palma de Mallorca, IllesBalears, Spain. E-mail: toni.frontera@uib.es
cSchool of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD, UK
dDepartment of Inorganic Chemistry, University of Barcelona, MartíiFranquès 1-11, 08028 Barcelona, Spain
eCentre for Healthcare Science and Technology, Indian Institute of Engineering Science and Technology, Shibpur, Howrah 711 103, India
First published on 5th September 2016
Abstract
A binuclear Cu(II) complex of formula [Cu(L1Hpy)Cl]2(ClO4)2 (1), where L1H2 is a new tridentate ligand, formed by condensation of 2-aminomethyl pyridine and pyridoxal (one of the forms of vitamin B6), has been synthesized. X-ray crystal structure determination shows that in this complex two Cu(II) ions are interconnected by complementary hydroxymethyl bridges of the two pyridoxal moieties, which is a very rare example in the literature. However, with a Cu⋯Cu separation of 6.574(1) Å and Cu–O(H)CH2– distance of 2.289 Å, the bridge is very weak, and DFT calculations, as well as ESI-MS data and solution spectral studies indicate that in a MeOH solution the complex exists predominantly as a mixture of monomers [Cu(L1Hpy)Cl]+ and [Cu(L1Hpy)(MeOH)]2+ with the former being the predominant form. The DFT calculations as well as EPR spectra suggest that the SOMO is a metal dx2−y2 orbital. The complex shows highly efficient catecholase activity with kcat = 3·46 × 105 h−1 and kcat/KM = 1.00 × 108 M−1 h−1, which are the best values reported in the literature, so far, for catecholase mimicking model complexes. DFT calculations show that the reduction of the Cu(II)/Cu(I) by the coordinated catechol and the resultant structural changes is the rate determining step in the catalytic cycle. The complex also binds DNA quite strongly with a binding constant of ∼105 M−1. DFT calculations suggest that the most probable binding mode of the complex is intercalation of the pyridine ring of the complex between two adenine or adenine and cytosine base pairs. The complex shows low cytotoxicity towards HCT and HeLa cells, though cytotoxicity towards the latter cell line is much more than the former. It was also found that the complex can be used as a fluorescence probe for imaging HCT cells.
Introduction
Catechol oxidase (CO) is an enzyme with a type-3 copper centre as its active site which catalyzes the oxidation of a wide range of o-diphenols (catechols), such as caffeic acid and its derivatives, to the corresponding o-quinones in a process known as catecholase activity.1 The o-quinones so formed are highly reactive compounds that can undergo auto-polymerization leading to the formation of a brown polyphenolic pigment, i.e. melanin, a process thought to protect damaged tissues against pathogens or insects.2 The ability of dicopper complexes to show catecholase activity is well-known and depends upon various factors such as metal–metal distances, Cu(II)/Cu(I) potentials of the complexes, the ligand structure, pH of the medium, the nature of the solvent and the number and nature of the bridging ligands.3–24 It is well established that dinuclear Cu(II) complexes with Cu⋯Cu distance of 2.9–3.2 Å show highest catecholase activity because of the requirement of a steric match between the substrate and catalyst.25 Recently, it was established that the presence of a positive charge centre close to the metal centre may enhance the catalytic activity.26Biological study of transition metal complexes, particularly their cytotoxicity studies and exploring the nature and strength of their DNA-binding ability is now receiving great attention. The objective of such studies is (a) search for new metallo-pharmaceuticals with greater efficacy than the conventional medicines (b) understanding the toxicity of metal complexes, particularly of those metal ions which are encountered in daily life. Moreover, as metal complexes can be easily studied by variety of spectroscopic as well as electrochemical techniques, so metal complex–DNA assembly has the potential application for DNA foot printing agents.27
In continuation of our studies on metal complexes of pyridoxal Schiff bases28–31 and exploring their bio-relevant applications we describe here the chemistry of a Cu(II) complex with a tridentate N2O-donor Schiff base of pyridoxal. We also investigated the catecholase activity, DNA binding property and cytotoxicity of the newly synthesized complex.
Experimental section
Materials and methods
Pyridoxal hydrochloride, 2-picolylamine, Cu(ClO4)2·6H2O were purchased from Aldrich. All other chemicals and solvents were of reagent grade and used as such. Solvents for spectroscopic and cyclic voltammetry studies were of HPLC grade obtained from Merck or Aldrich. Elemental analyses were performed on a Perkin-Elmer 2400 C, H, N analyzer. Infrared spectra were recorded as KBr pellets on a JASCO FT-IR-460 spectrophotometer. UV-vis spectra were recorded using a JASCO V-530 spectrophotometer, while fluorescence spectra at room temperature were recorded using PTI made QuantaMaster40 spectro-fluoremeter. Cyclic voltammograms were recorded in MeCN solutions, containing 0.1 (M) TEAP as supporting electrolyte, using a CH1120A potentiostat, with glassy carbon (GC) working electrode, Pt wire as counter electrode and Ag/AgCl/saturated KCl as reference electrode. The ferrocene/ferrocenium couple was observed at E0(ΔEp) = 0.48 V (100 mV) under our experimental conditions.Synthesis of [Cu(L1Hpy)Cl]2(ClO4)2 (1)
The tridentate Schiff base L1H2 (Scheme 1) was synthesized in situ by refluxing pyridoxal hydrochloride (0.406 g, 2 mmol) with 2-picolylamine (0.216 g, 2 mmol) in methanol. To it a methanolic solution of Cu(ClO4)2·6H2O (0.738 g, 2 mmol) was then added with constant stirring (Scheme 2). Deep green single crystals, suitable for X-ray diffraction, were obtained after two days by slow evaporation of the filtrate at room temperature. | ||
| Scheme 1 | ||
 | ||
| Scheme 2 Schematic representation of the template synthesis of the complex 1. | ||
Yield: 1.385 g (76%). Anal. calc. for C28H30Cl4Cu2N6O12: C, 36.89; H, 3.32; N, 9.22. Found: C, 36.80; H, 3.35; N, 9.30%. MS: m/z: 319.19 [CuL1Hpy]+ (100%)], (Fig. S1 in the ESI†), 638.52 [Cu2(L1)2]+, 675.47 [Cu2(L1Hpy)2Cl]+ (Fig. S2 in the ESI†). Electronic spectrum in MeOH solution λmax/nm (εmax/M−1 cm−1): 230 (49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 508), 270 (28099), 391 (13367), 650 (101) (Fig. 2). Selected IR bands (cm−1): 3318 (νO–H), 3095 (νN–H), 1637 (νC
508), 270 (28099), 391 (13367), 650 (101) (Fig. 2). Selected IR bands (cm−1): 3318 (νO–H), 3095 (νN–H), 1637 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1092 (νClO4−), 1019 (νC–O) (Fig. S4 in the ESI†).
N), 1092 (νClO4−), 1019 (νC–O) (Fig. S4 in the ESI†).
X-ray crystallography
Data for the complex was collected on a GV1000 Atlas four circle diffractometer by ω-scan method, at 150(2) K, using a graphite-monochromated Cu-Kα radiation. The data were solved by direct method and refined by full matrix least squares on F2 using SHELX 97.32 The non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed at calculated positions and refined as riding atoms using isotropic displacement parameters. Corrections for Lorentz polarization effects and semiempirical absorptions were applied.Density functional theory (DFT) calculations
The energies of all complexes included in this study were computed at the BP86-D3/def2-TZVP level of theory by using the program TURBOMOLE 7.0.33 The interaction energies were calculated with correction for the basis set superposition error (BSSE) by using the Boys–Bernardi counterpoise technique.34 For the calculations we have used the DFT-D functional with the latest available correction for dispersion (D3).35 In order to reproduce solvent effects, we have used the conductor-like screening model COSMO,36 which is a variant of the dielectric continuum solvation models. We have used methanol as solvent.37Catalytic oxidation of 3,5-DTBCH2
The catechol oxidase activity of the Cu(II) complex was examined using 3,5-ditertiarybutylcatechol (3,5-DTBCH2) as the substrate because of the ease of the oxidation of 3,5-DTBCH2 to 3,5-ditertiarybutylquinone (3,5-DTBQ) which has characteristic absorption at about 400 nm, that can be used to monitor the reaction. A 10−5 M solution of 1 in methanol was treated with 100 equiv. of 3,5-DTBC in methanol under aerobic conditions at room temperature. The absorbance versus wavelength scan of this solution was recorded at regular time intervals of 1 min in the wavelength range of 300–600 nm.Kinetics study of catalytic oxidation of 3,5-DTBCH2 by 1
The kinetics for the oxidation of the substrate 3,5-DTBCH2 was determined by the initial rate method at 25 °C. The concentration of the substrate 3,5-DTBCH2 was always kept at least 100 times larger than that of the complex and the increase of the quinone (3,5-DTBQ) concentration was determined by measuring its absorbance at 400 nm. Solutions of the substrate, with concentrations ranging from 0.001 to 0.04 M, were prepared from a concentrated stock solution in methanol. To a 1 cm spectrophotometer cell 2 mL of the substrate solution was poured. Then 40 μL of 10−3 M solution of 1 was quickly added to it so that the ultimate concentration of 1 became 2 × 10−5 M.Detection of hydrogen peroxide in the catalytic reaction
The formation of H2O2 during the catalytic reaction was detected by following the development of the characteristic band for I3− by spectrophotometry (λmax = 353 nm; ε = 26000 M−1 cm−1), upon reaction with I− with the reaction mixture. The oxidation reactions of 3,5-DTBCH2 in presence of 1 was carried out as in the kinetic experiments [compound] = 2.5 × 10−5 M; [3,5-DTBC] = 50 × 10−5 M, and after 1 h, the reaction was quenched by adding an equal volume of H2SO4 (20 × 10−3 M). The quinone formed was extracted three times with CH2Cl2. One milliliter of 10% aqueous KI solution was added to the aqueous layer. In order to accelerate the formation of I3− in the presence of H2O2, ammonium molybdate (3%) solution was added in a catalytic amount. In the presence of H2O2, a strong band at 350 nm, due to the formation of I3−, developed in electronic spectra. Since atmospheric dioxygen can oxidize I−, blank experiments (without catalyst or without 3,5-DTBCH2) were also performed. For the catalytic reactions performed in the presence of compound 1, the amount of H2O2 formed corresponds to 82% of the calculated value. However, only very minor formation of the I3− band was observed during the blank test.
Spectroscopic studies of interaction of 1 with DNA
:1, indicating that the DNA was sufficiently free from protein.38 The DNA concentration was determined by absorption spectroscopy using the molar extinction coefficient (6600 M−1 cm−1) at 260 nm.39 The intrinsic binding constant Kb for the interaction of the Cu(II) complex with DNA has been calculated from the absorption spectral titration data using eqn (1),40| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) | (1) |
Biological studies
:1 v/v ratio and incubated at 37 °C for 1 h in a CO2 incubator and observed under Andor spinning disk confocal microscope with excitation at 405 nm. Images were recorded at emission wavelength between 420 and 540 nm. After each treatment cells were washed three times with phosphate buffered saline (pH 7.4) to remove excess probe. Images were captured using EMCCD camera. Before microscopic imaging, all the solutions were aspirated and mounted on slides in a mounting medium containing Hoechst (1 μg mL−1) and stored in dark.Results and discussion
Synthesis and characterization of [Cu(L1Hpy)Cl]2(ClO4)2 (1) in the solid state
The ligand (L1H2) was prepared in situ by 1:1 condensation of pyridoxal hydrochloride with 2-picolylamine in methanol. The complex was synthesized by allowing the in situ prepared Schiff base to react with Cu(ClO4)2·6H2O in a methanol solution. Apart from the tridentate Schiff base ligand, which is protonated at the pyridine nitrogen, there is a coordinated chloride and a perchlorate anion that balance the charge of the complex species, but the axial positions of the metal ion remains vacant. In such a situation, oxygen atom of hydroxymethyl group shows unusual bridging property forming a rather long axial Cu–O bond resulting in a dinuclear complex in the solid state (Scheme 2). The participation of hydroxymethyl group in coordination is quite a rare occurrence and only three such examples, two involving Cu(II) complexes and one involving a Ni(II) complex, is known in the literature.41 The presence of ionic perchlorate and protonated pyridoxal-N can be detected in the infrared spectra of the complex with bands at 1091 and 3095 cm−1 respectively (see ESI† for details). The structure of the compound was determined by single crystal X-ray diffraction technique (Table 1).
| Formula | C28H30Cl4Cu2N6O12 |
| Formula weight | 911.46 |
| Crystal size (mm3) | 0.2632 × 0.1953 × 0.1253 |
| Temperature | 150(2) K |
| Diffraction wavelength (Å) | Cu-Kα (1.54184) |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 7.7545(13) |
| b (Å) | 8.7537(13) |
| c (Å) | 13.9700(11) |
| α (°) | 75.507(10) |
| β (°) | 83.103(10) |
| γ (°) | 65.615(15) |
| dcal | 1.810 |
| Z | 1 |
| μ (mm−1) | 5.158 |
| F(000) | 462 |
| Total reflections | 5562 |
| Unique reflections | 3245 |
| Observed data [I > 2σ(I)] | 2926 |
| Data/parameters/restraints | 2926/243/0 |
| R (int) | 0.0716 |
| R1, wR2 [I > 2σ(I)] | 0.0572, 0.154 |
| R1, wR2 (all data) | 0.0610, 0.159 |
| Goodness of fit | 1.04 |
The structural representation of the cationic unit of 1 is shown in Fig. 1 and important bond lengths and bond angles are listed in Table 2. In the dimer, each Cu(II) atom is equatorially bonded to phenoxide O, imine N and pyridine N atom of the ligand L1Hpy; in addition there is a coordinated chloride to complete the coordination requirement of the equatorial position. The hydroxymethyl oxygen atom of each ligand participates in a complementary axial coordination to the second Cu(II) atom of the dimer resulting in a square pyramidal coordination geometry around the Cu(II) centers (Addison parameter42 τ = 0.09). The equatorial Cu–O distance is shorter than the two Cu–N distances and all the equatorial bond lengths are within the normal range.43,44 The axial Cu–O bond is rather long at 2.289(2) Å, and hence the dimer may not be stable in solution (vide infra).
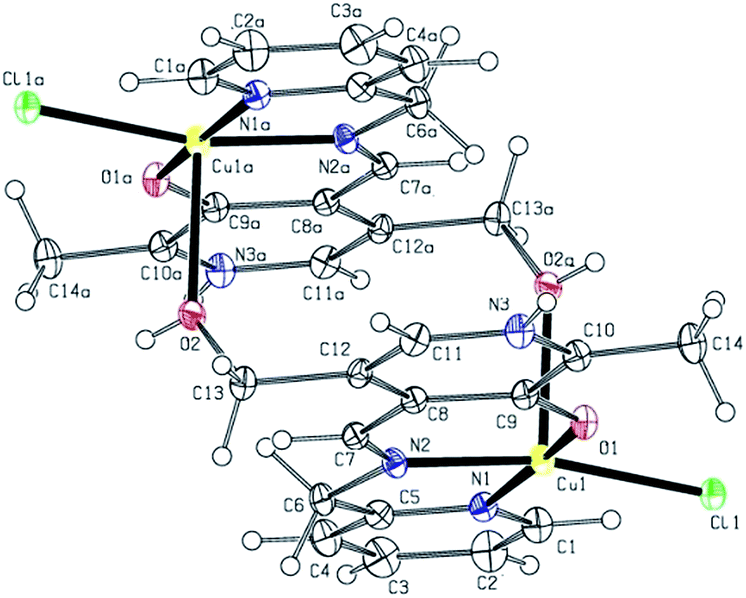 | ||
| Fig. 1 X-ray crystal structure of [Cu(L1Hpy)Cl]22+. For clarity the anions are omitted in the figure. | ||
| Bond lengths/Å | Bond angles/° | ||
|---|---|---|---|
| a Symmetry code for a is 1 − x, 1 − y, 1 − z. | |||
| Cu1–O1 | 1.929(2) | N2–Cu1–O2a | 96.70(11) |
| Cu1–N2 | 1.975(3) | N2–Cu1–N1 | 82.55(12) |
| Cu1–N1 | 2.007(3) | N2–Cu1–Cl1 | 167.46(9) |
| Cu1–Cl1 | 2.288(1) | N1–Cu1–O2a | 95.13(10) |
| Cu1–O2a | 2.289(2) | N1–Cu1–Cl1 | 95.91(9) |
| Cl1–Cu1–O2a | 95.83(7) | ||
| O1–Cu1–O2a | 88.59(10) | ||
| O1–Cu1–N2 | 90.99(11) | ||
| O1–Cu1–N1 | 72.87(11) | ||
| O1–Cu1–Cl1 | 89.75(8) | ||
The pyridine nitrogen atom (N3) of the pyridoxal ring remains protonated and to balance the charge there are two perchlorate ions per dimer. The H-bonding network and the resultant supramolecular structure are discussed in ESI.†
Characterization of the complex in solution
The ESI-MS spectrum of the complex in MeOH indicates that the predominant species in solution is a mononuclear entity, which is quite expected in view of the extreme weak nature of the bridge between the two metal centres in the binuclear complex in the solid state. DFT calculations suggest that the monomer is more stable by 3.0 kcal mol−1 in solution than the dimer. In methanolic solution, in the visible region of the electronic spectrum, there is a strong band at 391 nm, followed by a very weak absorption at 650 nm (Fig. 2). The electronic spectrum remains practically unchanged if small amount of perchloric acid is added to the solution (Fig. S6†), however, addition of alcoholic KOH alters the electronic spectrum, particularly the 391 nm band gradually disappears (Fig. S7†). On addition of excess of tetraethylammonium chloride (TEACl) to the methanolic solution of compound 1, the band positions remain unaltered, but the band intensities increase, particularly the band at 391 nm undergoes approximately 42% increase in intensity (Fig. S8†). Based on the above results we conclude that in MeOH solution the complex remains predominantly as [Cu(L1Hpy)Cl]+, with the pyridine nitrogen remaining protonated. A fraction of the complex (around 40%) undergoes solvolysis to form [Cu(L1Hpy)(MeOH)]2+, which in presence of excess TEACl is reconverted to [Cu(L1Hpy)Cl]+. The fact that band at 391 nm increases in intensity appreciably on addition of TEACl, but on addition of KOH, which not only deprotonates the pyridoxal nitrogen but displaces Cl− by OH− in the coordination sphere, the band disappears strongly suggest that band may be assigned to Cl− → Cu(II) charge transfer transition,45 though there may be some contribution from the phenolate O− → Cu(II) CT transition as well.46 The free ligand or its deprotonated forms produced by in situ addition of KOH solution show only weak absorption in this region and contribute only in a minor way towards the total intensity (Fig. S9†). The weak band at 650 nm is assigned to a d–d transition of a square planar or square pyramidal Cu(II) ion.47 The very strong bands at 230 and 270 nm are assigned to intraligand transitions. | ||
| Fig. 2 Electronic spectrum of compound 1 in methanol at 298 K. The d–d bands are shown in the inset. | ||
The conclusion derived from electronic spectra about the nature of the species in MeOH solution, is well supported by the EPR spectrum of the complex (Fig. 3) and conductivity measurements (for details of both see ESI†). The EPR spectrum in MeOH at 77 K is best simulated assuming two species Cu1 and Cu2 present in 3:2 ratio with the characteristic parameters for them being Cu1: g⊥ = 2.13, g∥ = 2.40, A∥ = 140 × 10−4 cm−1; Cu2: g⊥ = 2.10, g∥ = 2.30, A∥ = 107 × 10−4 cm−1. This compares well with the conclusion drawn from the electronic spectra where similar ratio of [Cu(L1Hpy)Cl]+ and [Cu(L1Hpy)(MeOH)]2+ was deducted in MeOH solution.
 | ||
| Fig. 3 X-band EPR spectra of compound 1 in methanol at 77 K (black line) and the simulated spectrum (line width 2.3 mT) (red line). | ||
Kinetic study of the catalytic reaction
The observed increase in absorbance at 400 nm during catalytic oxidation of 3,5-DTBCH2 by 1 is shown in Fig. 4. | ||
| Fig. 4 Increase in the absorbance after the addition of 100 equiv. of 3,5-DTBCH2 to a methanolic solution containing compound 1 (10−5 M). The spectra were recorded every 1 min. | ||
Since initial rate method follows saturation kinetics at higher substrate concentration we choose Michaelis–Menten model which was developed for enzyme kinetics and its linearized version in the form of a Lineweaver–Burk plot to calculate various kinetic parameters. The values of the Michaelis binding constant (KM), maximum velocity (Vmax), rate constant for the dissociation of the substrate (i.e., turnover frequency, kcat) were calculated for compound 1 from the Lineweaver–Burk graph 1/V vs. 1/[S] (Fig. 5) using the equation 1/V = {Km/Vmax}{1/[S]} + 1/Vmax. The kinetics data are listed in Table 3.
 | ||
| Fig. 5 Plot of the initial rate versus substrate concentrations for the oxidation reaction catalyzed by complex 1. The inset shows the Lineweaver–Burk plot. | ||
| Compound | Wavelength (nm) | Vmax (M min−1) | KM (M) | kcat (h−1) |
|---|---|---|---|---|
| 1 | 400 | 0.115 | 3.45 × 10−3 | 3.46 × 105 |
To get a better understanding of the complex–substrate intermediate and a mechanistic inference of catecholase activity during the oxidation reaction, we have recorded ESI-MS+ spectra of the mixture of the compound 1 and 3,5-DTBCH2 within 10 min of mixing in methanol. The compound 1 shows the base peak at m/z = 319.9 (100%) which can be assigned to a mononuclear species [CuL1Hpy]+. After addition of 3,5-DTBCH2 a new peak at 539.14 (Fig. S3†) indicates the formation of species [CuL1Hpy(3,5-DTBCH)].
Comparing the kinetic data for the compound 1 reported here, which is given in Table 3, with some of the most efficient Cu(II) catalysts of catechol oxidation reaction in MeOH medium, known so far (Table S2†), we can conclude that the Cu(II) catalyst reported in this work is the most efficient synthetic complex for the catecholase reaction. Our complex shows much more activity compared to the most efficient model catalyst reported to date (kcat = 3.24 × 104 h−1).1c
Theoretical study on the catecholase activity
To perform the theoretical study we have used the catalytic cycle proposed in Scheme 3 and we have optimized all intermediates at the BP86-D3/def2-TZVP level of theory. In the first step of the suggested mechanism (Scheme 3) the coordinated methanol molecule in (A) is replaced by the catechol. This coordination increases the acidity of the phenolic group. Moreover, we have observed in this optimized complex B an H-bond interaction between the other phenolic group of catechol and the chlorido ligand. We have indicated this interaction as a dashed line. The deprotonation of one of the phenolic groups leads to intermediate C. Interestingly this optimized complex shows that the Npy–Cu is elongated.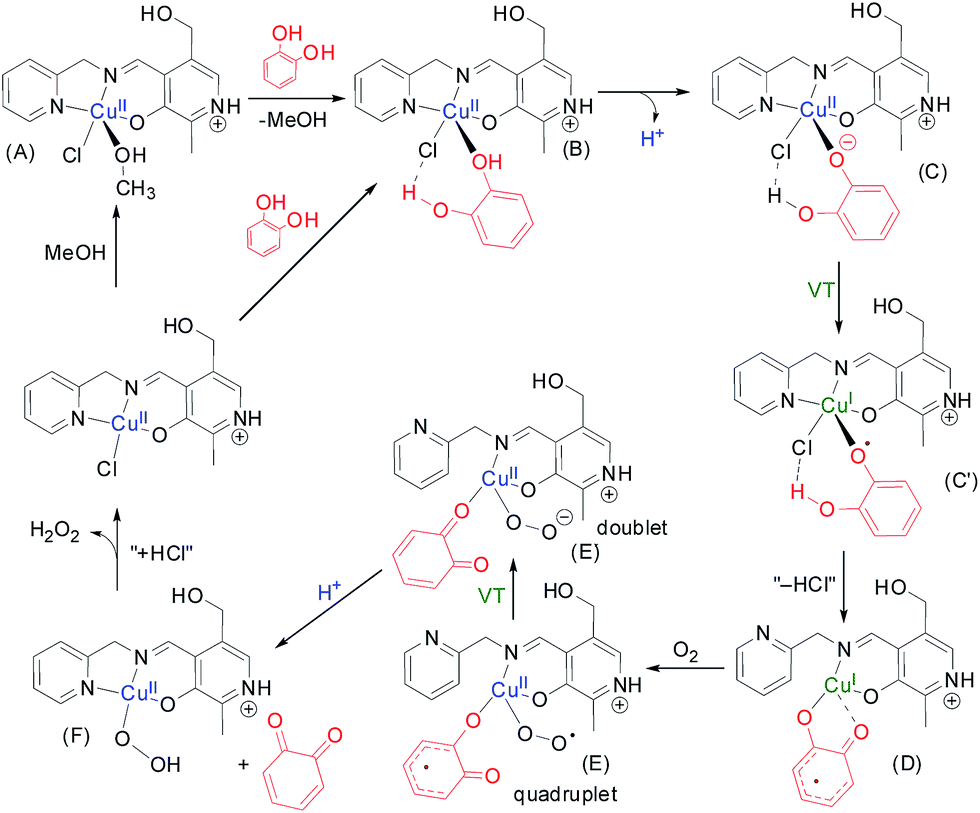 | ||
| Scheme 3 Catalytic cycle of catechol oxidation by a generic monocopper complex used in this study. One molecule of catechol is oxidized coupled with the reduction of molecular oxygen to hydrogen peroxide. | ||
The one electron transfer from the catechol moiety to the Cu takes place in this intermediate thus generating the semibenzoquinone with concomitant reduction of Cu(II) to Cu(I). At this point, we propose the elimination of “HCl” to yield compound D where the coordination of the semibenzo-o-quinone is bidentate. Interestingly, during the optimization of compound D, the pyridine moiety becomes uncoordinated. This facilitates the subsequent coordination of molecular oxygen yielding E, where the CuI is re-oxidized to CuII. At this point, the semibenzo-o-quinone is oxidized to o-quinone coupled with the formation of hydrogen peroxide (E′). The o-quinone is then released, facilitated by the re-coordination of the pyridine moiety of the organic ligand. Finally the chloride is coordinated to the Cu and the hydrogen peroxide is released to complete the catalytic cycle. We have also considered the possibility of intermediate D reacts with dioxygen via the semibenzo-o-quinone forming a C–O2˙ bond. However, the DFT calculations do not support this alternative route. The detection and quantitative estimation of hydrogen peroxide from the reaction mixture adds evidence in favour of the proposed mechanism.
The optimized geometries of the complexes A–G along with relative energies of transformation at each of the reaction steps are discussed in the ESI.† It was observed that the transformation of C to D has an energy cost of ΔEC→D = 28.6 kcal mol−1 at the PB86-D3/def2-TZVP level of theory in MeOH, and this is likely the rate determining step. The transformation of D to E involves the incorporation of molecular oxygen and the concomitant oxidation of the Cu(I) ion and this step in energetically favorable ΔED→E = −11.2 kcal mol−1.
We have also analyzed the spin density and single-occupied molecular orbitals (SOMOs) of intermediate E before (quadruplet) and after (doublet, denoted as E′ in Scheme 3) the electron transfer (see Fig. 6). In the quadruplet, the spin density plot shows that the spin density is delocalized in the metal center, the O2˙− and the semibenzo-o-quinonate ligand.
 | ||
| Fig. 6 Spin density and SOMOs of intermediates E and E′. | ||
The highest SOMO corresponds to the contribution of the π-system of the semiquinonate moiety with partial contribution of the d atomic orbitals of the metal centers. In contrast the π-system of catechol does not participate in the SOMO−1 and it has the contribution of the atomic orbitals of the metal center, the atoms directly bonded to it and the O2˙−. Upon the electron transfer (E′), it can be observed that the π-system of the benzo-o-quinone does not contribute to the SOMO, which has the contribution of the atomic orbitals of the metal center, the atoms directly bonded to it and the O22−. The energy difference between the optimized structures of E (quadruplet) and E′ (doublet) is only 0.63 kcal mol−1, therefore the electron transfer from the semibenzo-o-quinonate to the Cu/O2˙− is not a rate determining step.
DNA binding study
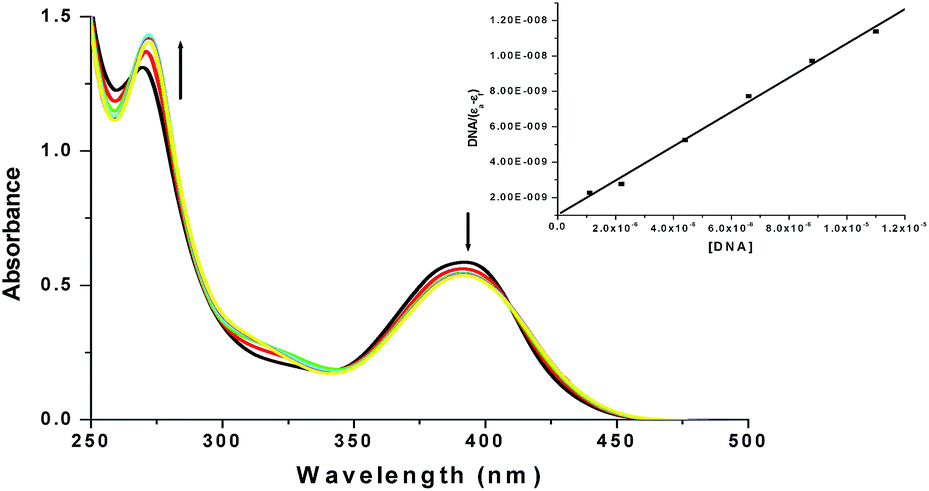 | ||
| Fig. 7 Absorption spectra of complex 1 in the absence and presence of increasing amount of CT-DNA at 25 °C in 5 mM aqueous Tris–HCl buffer at pH 7.4. Inset: plot of [DNA]/(εa−εf) vs. [DNA]. | ||
 | ||
| Fig. 8 Emission spectra of EB bound DNA with increasing amounts of 1. Arrow shows the intensity change upon increasing the complex concentration. | ||
The addition of 1 to DNA, pretreated with EB, causes an appreciable reduction in fluorescence intensity, indicating that complex 1 competes with EB to bind to DNA. The reduction of the emission intensity gives a measure of the DNA binding propensity of the complex and the stacking interaction (intercalation) between the adjacent DNA base pairs.50 The quenching constant K was determined from the classical Stern–Volmer equation51 I0/I = 1 + KSV [Q] where I0 and I are the fluorescence intensities in the absence and presence of the complex, respectively. KSV is the linear Stern–Volmer quenching constant, [Q] is the concentration of the Cu(II) (quencher) complex. The quenching of EB bound to DNA by complex 1 is in good agreement with the linear curve of Stern–Volmer equation (Fig. 9). From the slope of plot of I0/I versus [complex], the KSV value for the copper(II) complex was found to be 1.79 × 104 (R = 0.999), indicating a strong affinity of the copper(II) complex to CT-DNA.
 | ||
| Fig. 9 Plot of I0/I vs. [CuII complex]. | ||
The apparent binding constant (Kapp) value is determined from the equation
| Kapp × [complex]50 = KEB × [EB] |
We have considered A–T and G–B base pairs and computed the interaction energies of each pair with the complex in two different orientations. In Fig. 10 we show the optimized geometries of the complexes between the A–T pair with compound 1.
 | ||
| Fig. 10 PB86-D3/def2-TZVP optimized geometries and interaction energies of A–T base pair complexes with the monomeric form of 1. | ||
Moreover, we have also computed the interaction energies of the free ligand in order to investigate the influence of the metal center on the interaction energies. We have studied two different orientations where either the thymine (A) or adenine (B) establishes a π-stacking interaction with the pyridine moiety (in orange) of the ligand. In the first orientation (A), the A–T pair conserves its planarity and the interaction energy is ΔE1 = −28.1 kcal mol−1. In contrast, the other orientation affects the coplanarity of the bases and the interaction energy is larger (in absolute value, ΔE2 = −34.0 kcal mol−1). The interaction energies computed for the A–T complexes with the free ligand (see Fig. 10C and D) in any of both orientations are smaller than those using the Cu-complex, indicating that the metal center increases the ability of the ligand to bind the DNA base pair.
Similar calculations on interaction of complex 1 with the G–C pair show that the most favorable complex corresponds to the cytosine π-stacked with the pyridine moiety of the ligand (Fig. S15†) and the interaction energy is similar (ΔE5 = −32.1 kcal mol−1) to that obtained for the A–T complex (see Fig. 10B). Therefore, compound 1 has approximately the same ability to bind A–T and G–C pairs. Consequently, the best combinations for the intercalation of 1 to the DNA are either the pyridine moiety of the complex sandwiched between two adenine bases or between an adenine and a cytosine. The complexes of the G–C pair with the free ligand are less favorable than with the Cu-complex (see Fig. S15C and D†) in line with the previous result obtained for the A–T pair.
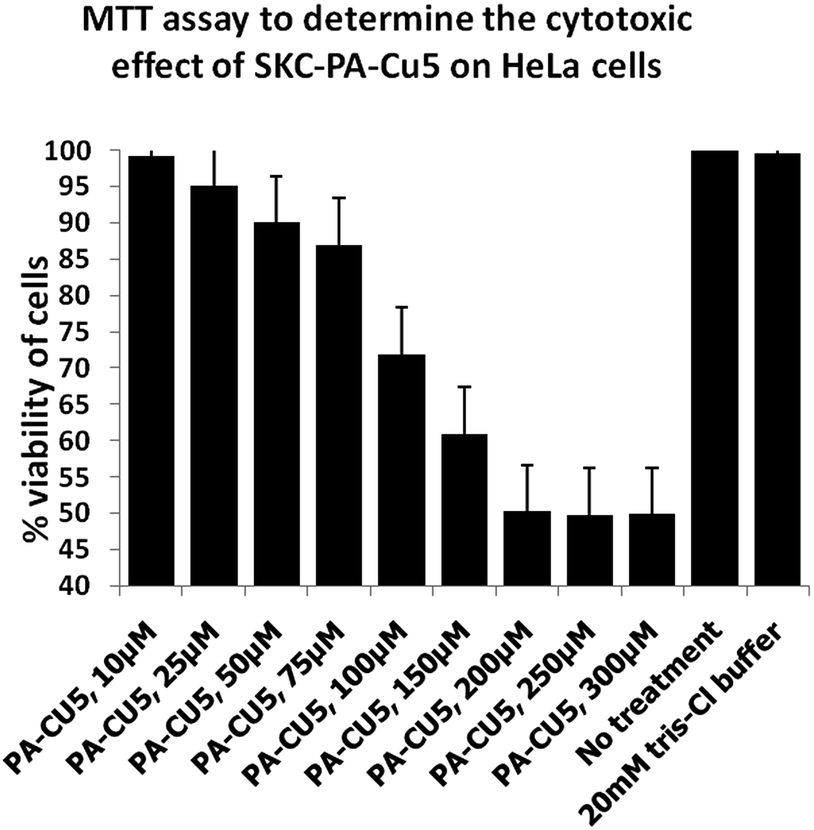 | ||
| Fig. 11 Cytotoxicity study of complex 1 (referred as SKC-PA-Cu5 in the diagram) on HeLa cells. | ||
The fluorescence images were recorded before and after the addition of compound 1 (20 μM). The compound can penetrate cells easily without any morphometric distortion in case of HCT cells (Fig. 12) but in case of HeLa cells after incubation the cells lost their morphological integrity (Fig. S17†). The cell membrane was destroyed and nucleus too came out in many cases, leaving the cytoplasmic debri all over the microscopic field which once again establishes that the compound is much more cytotoxic towards HeLa cells than HCT cells. For the HCT cells, cells incubated without the probe exhibited no fluorescence, whereas a bright fluorescence signal was observed in the cells stained with the complex, which shows that 1 is cell membrane permeable and can be efficiently used for biological imaging in very low dose.
 | ||
| Fig. 12 Confocal microscopic images of HCT 116 cells; all images were acquired with a 100 × objective lens. (a) Bright field image of cells (b) fluorescence images of cells without probe (complex 1), nuclei counterstained with Hoechst 33342 fluorescent stain (1 μg mL−1) (c) fluorescence images of cells with probe (complex 1), excited at 405 nm (d) overlay image of (b) and (c). | ||
Conclusions
We report here a rare hydroxymethyl–bridged dicopper complex of a pyridoxal based tridentate Schiff base ligand. DFT calculations show the corresponding mononuclear species is more stable than the binuclear complex in solution, which is supported by solution studies. The complex can act as very efficient catalyst for the oxidation of catechols to o-quinones and the kcat value of 1 for oxidation of 3,5-di-tert-butylcatechol (3,5-DTBCH2) to 3,5-di-tert-butylquinone (3,5-DTBQ) is ∼11 times higher than the most efficient catalyst reported till date.1c From electronic absorption and competitive fluorescence titration it is confirmed that the Cu(II) complex is also an avid binder of DNA. DFT calculations were used to understand the possible site of intercalation interaction of the complex with DNA and it was concluded that the complex binds DNA by the π-stacking interaction of the pyridine moiety of the complex with two adenine or an adenine and a cytosine base pairs. The complex proved to be mildly cytotoxic towards HCT and HeLa cell lines and it proved to be an excellent fluorescent probe for imaging HCT cells.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
P. A. and B. G. thanks UGC and IIEST, Shibpur, respectively for fellowships. S. K. C acknowledges financial grant from DST (West Bengal). A. B. and A. F. acknowledge financial support from the Spanish Ministry of Economy and Competitiveness (MINECO) through projects CTQ2014-57393-C2-1-P and CONSOLIDER INGENIO 2010 CSD2010-00065 (FEDER funds). M. C. acknowledge financial support from the Spanish Ministry of Economy and Competitiveness (MINECO) through project CTQ2015-63614-P.References
- (a) A. Rompel, H. Fischer, D. Meiwes, K. Büldt-Karentzopoulos, R. Dillinger, F. Tuczek, H. Witzel and B. Krebs, J. Biol. Inorg. Chem., 1999, 4, 56 CrossRef CAS PubMed; (b) A. Biswas, L. K. Das, G. B. Drew, D. C. Michael and A. Ghosh, Inorg. Chem., 2012, 51, 10111 CrossRef CAS PubMed; (c) K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083 CrossRef CAS PubMed.
- B. J. Dervall, Nature, 1961, 189, 311 CrossRef.
- (a) C. Eicken, F. Zippel, K. Büldt-Karentzopoulos and B. Krebs, FEBS Lett., 1998, 436, 293 CrossRef CAS PubMed; (b) R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211 CrossRef; (c) P. Gentschev, N. Moller and B. Krebs, Inorg. Chim. Acta, 2000, 300, 442 CrossRef; (d) M. Merkel, N. Moller, M. Piacenza, S. Grimme, A. Rompel and B. Krebs, Chem.–Eur. J., 2005, 11, 1201 CrossRef CAS PubMed.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553 CrossRef CAS PubMed.
- J. Manzur, A. M. Garcia, A. Vega and E. Spodine, Polyhedron, 1999, 18, 2399 CrossRef CAS.
- M. Gupta, P. Mathur and R. J. Butcher, Inorg. Chem., 2001, 40, 878 CrossRef CAS PubMed.
- N. N. Murthy, M. Mahroof-Tahir and K. D. Karlin, Inorg. Chem., 2001, 40, 628 CrossRef CAS PubMed.
- C. H. Kao, H. H. Wei, Y. H. Liu, G. H. Lee, Y. Wang and C. J. Lee, J. Inorg. Biochem., 2001, 84, 171 CrossRef CAS PubMed.
- C. Fernandes, A. Neves, A. J. Bortoluzzi, A. S. Mangrich, E. Rentschler, B. Szpoganicz and E. Schwingel, Inorg. Chim. Acta, 2001, 320, 12 CrossRef CAS.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429 CrossRef CAS.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788 CrossRef CAS PubMed.
- (a) J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem.–Eur. J., 2002, 8, 247 CrossRef CAS PubMed; (b) J. Ackermann, S. Buchler and F. Meyer, C. R. Chim., 2007, 10, 421 CrossRef CAS.
- O. Seneque, M. Campion, B. Douziech, M. Giorgi, E. Riviere, Y. Journaux, Y. L. Mest and O. Reinaud, Eur. J. Inorg. Chem., 2002, 2007 CrossRef CAS.
- (a) I. A. Koval, D. Pursche, A. F. Stassen, P. Gamez, B. Krebs and J. Reedjik, Eur. J. Inorg. Chem., 2003, 1669 CrossRef CAS; (b) I. A. Koval, M. Huisman, A. F. Stassen, P. Gamez, M. Lutz, A. L. Spek and J. Reedijk, Eur. J. Inorg. Chem., 2004, 591 CrossRef CAS.
- B. Sreenivasulu, M. Vetrichelvan, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2005, 4635 CrossRef CAS.
- R. Wegner, M. Gottschaldt, P. Wolfgang, E.-G. Jager and D. Klemm, J. Mol. Catal. A: Chem., 2003, 201, 93 CrossRef CAS.
- M. Thirumavalavan, P. Akilan, M. Kandaswamy, K. Chinnakali, G. S. Kumar and H. K. Fun, Inorg. Chem., 2003, 42, 3308 CrossRef CAS PubMed.
- J. Kaizer, R. Csonka, G. Speir, M. Giorgi and M. Reglier, J. Mol. Catal. A: Chem., 2005, 235, 81 CrossRef CAS.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083 CrossRef CAS PubMed.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS PubMed.
- V. K. Bhardwaj, N. Aliaga-Alcalde, M. Corbella and G. Hundal, Inorg. Chim. Acta, 2010, 363, 97 CrossRef CAS.
- I. A. Koval, K. Selmeczi, C. Belle, C. Philouze, E. Saint-Aman, I. Gautier- Luneau, A. M. Schuitema, M. v. Vliet, P. Gamez, O. Roubeau, M. Luken, B. Krebs, M. Lutz, A. L. Spek, J.-L. Pierre and J. Reedijk, Chem.–Eur. J., 2006, 12, 6138 CrossRef CAS PubMed.
- R. E. H. M. B. Osório, R. A. Peralta, A. J. Bortoluzzi, V. R. deAlmeida, B. Szpoganicz, F. L. Fischer, H. Terenzi, A. S. Mangrich, K. M. Mantovani, D. E. C. Ferreira, W. R. Rocha, W. Haase, Z. Tomkowicz, A. dos Anjos and A. Neves, Inorg. Chem., 2012, 51, 1569 CrossRef PubMed.
- E. Mijangos, J. Reedijk and L. Gasque, Dalton Trans., 2008, 1857 RSC.
- N. Oishi, Y. Nishida, K. Ida and S. Kida, Bull. Chem. Soc. Jpn., 1980, 53, 2847 CrossRef CAS.
- J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauzá, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442 CrossRef CAS PubMed.
- (a) G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. Garcia, Coord. Chem. Rev., 2013, 257, 2848 CrossRef CAS; (b) B. M. Zeglis, V. C. Pieree and J. K. Barton, Chem. Commun., 2007, 4565 RSC; (c) K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS PubMed; (d) D. Desbouis, I. P. Troitsky, M. J. Beleousoff, L. spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897 CrossRef CAS.
- C. Das, P. Adak, S. Mondal, R. Sekiya, R. Kuroda, S. I. Gorelsky and S. K. Chattopadhyay, Inorg. Chem., 2014, 53, 11426 CrossRef CAS PubMed.
- S. Naskar, S. Naskar, H. Mayer-Figge, W. S. Sheldrick and S. K. Chattopadhyay, Polyhedron, 2011, 30, 529 CrossRef CAS.
- S. Naskar, S. Naskar, R. J. Butcher and S. K. Chattopadhyay, Inorg. Chim. Acta, 2010, 363, 404 CrossRef CAS.
- S. Mondal, P. Adak, C. Das, S. Naskar, B. Pakhira, A. L. Rheingold, E. Sinn, C. S. Eribal and S. K. Chattopadhyay, Polyhedron, 2014, 81, 428 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Hacer, H. Horn and C. Kömel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- S. B. Boys and F. Bernardy, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- A. Klampt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699 CrossRef.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1993, 32, 2573 CrossRef CAS PubMed.
- J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081 CrossRef CAS.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051 CrossRef CAS.
- (a) V. M. Leovac, V. S. Jevtovic, L. Jovannic and G. A. Bogdamovic, J. Serb. Chem. Soc., 2005, 70, 393 CrossRef CAS; (b) M. Belicchi-Ferrari, F. Bisceglie, C. Casoli, S. Durot, I. Morgenstern-Badarau, G. Pelosi, E. Pilotti, S. Pinelli and P. Tarasconi, J. Med. Chem., 2005, 48, 1671 CrossRef CAS PubMed; (c) V. M. Leovac, S. Marković, V. Divjaković, K. M. Szécsényi, M. D. Joksović and I. Leban, Acta Chim. Slov., 2008, 55, 850 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, Dalton Trans., 1984, 1349 RSC.
- S. Mondal, S. Naskar, A. K. Dey, E. Sinn, C. Eribal, S. R. Herron and S. K. Chattopadhyay, Inorg. Chim. Acta, 2013, 398, 98 CrossRef CAS.
- S. Naskar, S. Naskar, H. Mayer-Figge, W. S. Sheldrick, M. Corbella, J. Tercero and S. K. Chattopadhyay, Polyhedron, 2012, 35, 77 CrossRef CAS.
- (a) R. Valiente, F. Rodríguez, M. Moreno and M. Hitchman, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 265, 176 CrossRef CAS; (b) V. K. Miskowski, J. A. Thich, R. Solomon and H. J. Schugar, J. Am. Chem. Soc., 1976, 98, 8344 CrossRef CAS.
- (a) S. Ghosh, J. Cirera, M. A. Vance, T. Ono, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2008, 130, 16262 CrossRef CAS PubMed; (b) B. A. Jazdzewski and W. B. Tolman, Coord. Chem. Rev., 2000, 200–202, 633 CrossRef CAS; (c) K. D. Karlin, J. C. Hayes, Y. Gultneh, R. W. Cruse, J. W. McKown, J. P. Hutchinson and J. Zubieta, J. Am. Chem. Soc., 1984, 106, 2121 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1984. p. 553 Search PubMed.
- E. Nyarko, N. Hanada, A. Habib and M. Tabata, Inorg. Chim. Acta, 2004, 357, 739 CrossRef CAS.
- J. B. Lepecq and C. Paoletti, J. Mol. Biol., 1967, 27, 87 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237 CrossRef CAS PubMed.
- P. X. Xi, Z. H. Xu, X. H. Liu, F. J. Chen and Z. Z. Zeng, Chem. Pharm. Bull., 2008, 56, 541 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data in CIF format, IR spectra, mass of the complex, FMOs of the monomer [Cu(L1Hpy)Cl(MeOH)]+, H-bonding network and cyclic voltammogram of 1. CCDC 1465420. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14059a |
| This journal is © The Royal Society of Chemistry 2016 |