
Investigation on polyvinyl-alcohol-based rapidly gelling hydrogels for containment of hazardous chemicals†
Abstract
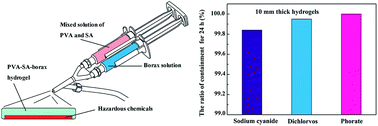
The specific problem that is of concern here is to stop or at least significantly retard the diffusion of hazardous chemicals which are accidentally released into public places. The solution proposed is to employ rapidly gelling hydrogels that display an effective barrier property to cover the hazardous chemicals. Rapidly gelling hydrogels were prepared by blending a solution of polyvinyl alcohol (PVA) and borax. Sodium alginate (SA) was incorporated so as to improve the stretchability of the PVA–borax crosslinked system. The PVA–SA–borax hydrogels with PVA contents of 15 wt%, SA contents of 0.6 wt%, and borax : PVA ratios of 1 : 25 can be formed within 1 minute, which exhibited self-healing capability and favorable thermal adaptability. The barrier properties of the PVA–SA–borax hydrogels to sodium cyanide, dichlorvos and phorate were investigated, and more than 99% of the sodium cyanide, dichlorvos and phorate can be confined for 24 h by using 10 mm thick hydrogels.
 Please wait while we load your content...
Please wait while we load your content...

