DOI:
10.1039/C6RA13908F
(Paper)
RSC Adv., 2016,
6, 78090-78099
Modified wollastonite sequestrating CO2 and exploratory application of the carbonation products†
Received
29th May 2016
, Accepted 27th July 2016
First published on 1st August 2016
Abstract
The feasibility of wollastonite carbonation for CO2 sequestration was systematically investigated, and the recovery of the by-products (SiO2, NH4Cl) in the carbonation process, the controllable preparation of carbonation product (CaCO3) with different polymorph and morphology during the carbonation process, and the application of the carbonation product were evaluated. The ammonia dosage and volume of the Ca2+ leaching solution showed a prominent effect on the wollastonite carbonation ratio. The synthesis of the carbonation product (CaCO3) with different polymorphs and morphologies was successfully realized by controlling the temperature and ammonia dosage during the carbonation process. A reaction mechanism of wollastonite carbonation was also proposed by thermodynamic research of the gas–liquid–solid reaction. The carbonation ratio could reach up to 93%, and 1 ton wollastonite could sequester 350 kg of CO2 under optimized conditions. The crystalline phase of the as-obtained CaCO3 was spherical vaterite and cubic calcite, which could meet the relevant standards for the industrial precipitated calcium carbonate CaCO3 (HG/T 2226-2010). The physical properties of the corresponding PP/CaCO3 composites were very close. The cost of wollastonite carbonation was estimated to be 120 $ per t without industrial scale experiment. The wollastonite carbonation strategy showed potential application for CO2 sequestration.
1. Introduction
Human beings have played a very important role in controlling the global environment over the past century.1 Modernization and industrialization of countries have been changing our environmental conditions. Undoubtedly, energy has played a very important role in the course of human development.2 The consumption of fossil fuels was accompanied by a large amount of carbon dioxide emissions.3–5 According to the Emission Database for Global Atmospheric Research,6 excessive fossil fuel consumption has increased the global CO2 emission to 36.1 billion tonnes in 2013, which is 48% more than that of two decades ago. Over the past century, atmospheric CO2 level increased more than 39%, from 280 ppm during pre-industrial times to the record high level of 400 ppm in May 2013 with a corresponding increase in global surface temperature of about 0.8 °C.7 The global carbon cycle cannot absorb all the anthropogenically produced carbon dioxide in the coming centuries, so adaptation technologies are urgently required. There are two main strategies for lowering the atmospheric concentration of CO2: developing a clean energy and sequestering CO2. Owing to the high cost of other energy sources, societal pressure and the established infrastructure of countries, fossil fuels will remain as the world's primary energy sources for the foreseeable future.8–10 Methods for sequestering CO2 currently being considered by industrialized countries include the enhancement of terrestrial carbon sinks as well as geological ocean and mineral sequestration.11–15
Mineral carbonation, due to its rich in raw material, has recently been considered as a leading route for CO2 sequestration.16–19 Yan et al.20 systematically studied the mineral carbonation reaction characteristics of wollastonite, serpentine and olivine under low-medium pressure conditions. At T = 150 °C, P = 40 bar and particle sizes <30 μm, the highest carbonation conversion efficiency of 83.5%, 47.7% and 16.9% was achieved for wollastonite, serpentine and olivine, respectively. By developing an attrition-leaching hybrid process for direct aqueous mineral carbonation, Julcour et al.21 reported that the carbonation yield for olivine and serpentinised ores could reach up to 35% in 5 h, 80% in 24 h in water, and 70% in 5 h with inorganic additives. Vance et al.22 discussed the carbonation of portlandite by carbon dioxide in the liquid and supercritical states as a potential route toward CO2-neutral cementation. It is only slightly sensitive to the effects of temperature, pressure, and the state of CO2 over the range of 6 MPa ≤ P ≤ 10 MPa and 8 °C ≤ T ≤ 42 °C. More than 80% carbonation of Ca(OH)2 was achieved in 2 h upon contact with liquid CO2 at ambient temperatures. During the Mg(OH)2 slurry carbonation, Fricker et al.23 investigated the different carbonate phases and their formation kinetics, and they found that the reaction temperature was a dominant parameter driving the formation of specific carbonate phases. Wang et al.24 examined the serpentine carbonation of different flue gases and maintained the pH in the system using ammonium salts. An average carbonation efficiency of about 90% was achieved at 80 °C after 30 min. Overall, previous studies have described the different mineral carbonation processes in detail, but the carbonation ratio so far is still rarely able to surpass 90%. The researchers focused on CO2 sequestration amount and carbonation ratio; there was little attention on the carbonation products. The cost of mineral carbonation for CO2 sequestration was still very high.
In this study, wollastonite was used in a carbonation experiment, where hydrochloric acid and ammonia were employed as the reaction media to speed up the reaction. Many researchers have reported wollastonite carbonation,25–32 where they all focused on how to improve the carbonation ratio. In this study, we investigated the feasibility of the route using an experimental approach and studied the entire procedure of wollastonite carbonation for CO2 sequestration, including acid-leaching of wollastonite, carbonation of wollastonite, recovering of byproducts and application of carbonation product. To broaden the application field of the carbonation product, polymorph and morphology controllable preparation of carbonation product during the carbonation process was discussed. Moreover, we explained the underlying mechanisms of the carbonation process by thermodynamics analysis, and the economic cost of wollastonite carbonation was also simply estimated.
2. Experimental
2.1. Materials preparation
Pristine wollastonite used in this study was obtained from Jiangxi, China. Detailed analysis of the raw material was in the previous study.25 Polypropylene pellets (PP, ρ = 0.91 g cm−3, ∼3 mm in size) was obtained from China Petroleum & Chemical Co., Ltd. The titanate coupling agent, hydrochloric acid, ethylalcohol and ammonia were used without further purification. Carbon dioxide was industry-grade with a purity of 99.9%.
From our previous research into acid-leaching for clay minerals,33–35 pristine wollastonite was first acid-leached at 80 °C for 120 min with a 6 M hydrochloric acid solution. 20 g of pristine wollastonite was placed into a 500 mL reaction vessel, and 200 mL of hydrochloric acid solution was then added. A magnetic stir bar was used to ensure adequate mixing throughout the reaction. A hot bath was used to maintain the desired reaction temperature. After the desired reaction time, the solid (SiO2) was then washed and dried for further analysis. The acid-leaching filtrate was neutralized using ammonia for the carbonation experiment. A carbonation experiment was performed by adding ammonia and carbon dioxide into the neutral acid-leaching filtrate. The CO2-rich gas was injected at a predetermined flow rate at normal pressure. A pH meter was installed to monitor the progress of the carbonation reaction. The pH value was recorded at 1 min interval until the pH was relatively constant. Carbonation experiment was referred to the desulfurization residue carbonation.36 After a desired reaction time, the mixture was cooled and filtered to separate the solids (CaCO3) from the solution. The solids were then washed with distilled water until they reached a neutral pH, before being dried in an oven at 110 °C for 24 h. The carbonation filtrate was evaporated at 80 °C to obtain NH4Cl for further analysis. The modification process and preparation of the PP/carbonation product composites are shown in ESI† in detail.
2.2. Characterization
The composition of the sample was determined by X-ray fluorescence (XRF) and chemical titration. Powder XRD measurements of the samples were conducted with a DX-2700 X-ray diffractometer using Cu Kα radiation (λ = 0.15406 nm) at a scanning ratio of 0.02 deg. s−1 with a voltage of 40 kV at 40 mA. SEM was performed using a JEOL JSM-6360LV scanning electron microanalyzer with an accelerating voltage of 5 kV. Fourier transform infrared (FTIR) spectroscopy of the samples over the range of 4000–400 cm−1 was performed on a Nicolet Nexus 670 FTIR spectrometer. The samples were ground with KBr crystals, and the mixture was pressed into a pellet for the FTIR measurements. The particle size was measured using a particle size analyzer (LS-POP(6), Zhuhai OMEC Instrument Co., Ltd, China). The physical properties of the additive-PP composites were measured using a mechanical testing machine (WDW-1000, China) and a softening temperature testing machine (XRW-300MA, China) according to the standards of GB/T 1042-1992 and GB/T 9341-2000, respectively. The calculation methods of the carbonation ratio (η) and the molar content (%) of CaCO3 polymorphs are described in ESI,† respectively.
3. Results and discussion
3.1. Wollastonite carbonation process
The effects of ammonia dosage, CO2 flow rate, volume of Ca2+ leaching solution and temperature on the carbonation of wollastonite are discussed by monitoring the pH of the reaction solution and carbonation ratio. The detailed analysis is shown in Fig. S1–S4 in ESI.† According to the analysis, the ammonia dosage and the volume of Ca2+ leaching solution were the most important process variables influencing the wollastonite carbonation process. The effects of either the CO2 flow rate or temperature was not as notable as the ammonia dosage and volume of Ca2+ leaching solution. Under the optimized conditions (8 mL, 100 mL min−1, 150 mL, 30 °C), the carbonation ratio can reach about 93%.
3.2. Application of carbonation product (CaCO3)
The abovementioned research has clearly demonstrated the feasibility of using wollastonite for CO2 sequestration with industry-grade using the proposed scheme. By following the optimized process, the carbonation ratio (η) can reach 92.5%, and 1 ton wollastonite was able to sequester 350 kg of CO2, a value greater than the vast majority of the other wollastonite carbonation obtained previously (Table 1). Fig. 1 shows the XRD and SEM results of the acid-leached wollastonite, carbonation by-product and carbonation product obtained under the optional conditions. There was a prominent amorphous package of SiO2 in the acid-leached wollastonite (Fig. 1a), and the crystal phase was hydrogen silicate. The morphology of the particles was granular (Fig. 1c), and the average particle size (d50) was 21.8 μm (Fig. 1b). The content of SiO2 in the samples was 98.3%. The whiteness of the samples was 92%. The only crystal phase was sal ammoniac in the carbonation by-product (Fig. 1a). The purity of the NH4Cl byproduct evaluated by the XRD pattern was 99%, which could meet the relevant standards for the ammonium chloride NH4Cl (GB/T2946-2008). The main crystal phase of carbonation product (CaCO3) was of vaterite containing some calcite. The morphology of the particles was spherical and cubic (Fig. 1d), and the average particle size (d50) was 16.7 μm (Fig. 1b). The content of CaCO3 in the samples reached 99.1%. The whiteness of the samples was 93%. The performance indices of the carbonation product (CaCO3) met the relevant standards for the industrial precipitated calcium carbonate CaCO3 (HG/T 2226-2010); therefore, it could possibly replace calcium carbonate used in plastics field. In the following study, we experimentally analyzed the carbonation product (CaCO3) used in PP-packing in detail.
Table 1 Comparisons of CO2 sequestration by wollastonite carbonations
| No. |
Process conditions |
Carbonation ratio (%) |
References |
| 1 |
Carbonation at 80 °C and 3 MPa for 30 min |
20 |
27 |
| 2 |
Particle size < 106 μm, T = 100–225 °C for 15 min |
45 |
50 |
| 3 |
Carbonation at T = 185 °C, P = 150 atm for 1 h and particle sizes < 37 μm |
82 |
51 |
| 4 |
NaHCO3 was used as agent, carbonation at T = 150 °C, P = 40 bar and particle sizes < 30 μm |
83.5 |
20 |
| 5 |
150 μm < particle size < 250 μm after 2 days, T = 90 °C, pCO2 = 250 bar |
85 |
26 |
| 6 |
NH4OH was used as agent, carbonation at room temperature and atmospheric pressure |
91.1 |
25 |
| 7 |
Carbonation at T = 100 °C and P = 40 atm |
100 |
28 |
| 8 |
NH4OH was used as agent, carbonation at room temperature and atmospheric pressure |
92.5 |
This work |
 |
| | Fig. 1 (a) XRD patterns of the acid-leached product (SiO2), carbonation product (CaCO3), and by-product (NH4Cl) for mineral carbonation of wollastonite, (b) particle size distribution of SiO2, and SEM images of (c) SiO2 and (d) CaCO3. | |
CaCO3 was first surface-modified by the titanate coupling agent, and then added to polypropylene (PP) to prepare PP/CaCO3 composites. The detailed modification analysis is shown in Fig. S5.† The XRD patterns of PP and PP/modified CaCO3 composites are shown in Fig. 2. The reflections at 2θ 13.68°, 16.50°, 18.16°, and 21.34° were characteristic of α-PP crystalline phase (Fig. 2a), corresponding to crystal planes (110), (040), (130), and (041),37 respectively. PP/modified CaCO3 composites had similar diffraction patterns to pure PP, indicating that no other PP crystalline phases appeared. The results illustrated that the morphology of modified CaCO3 for fillers could not induce the crystalline phase of PP. The intensities of the calcium carbonate crystal diffraction reflections clearly increased with increasing modified CaCO3 powder content (Fig. 2a); however, this did not lead to a crystalline transformation of PP. Fig. 2b–f show the SEM images of the PP/modified CaCO3 composites, indicating that CaCO3 particles increased with the addition of modified CaCO3 powder content.
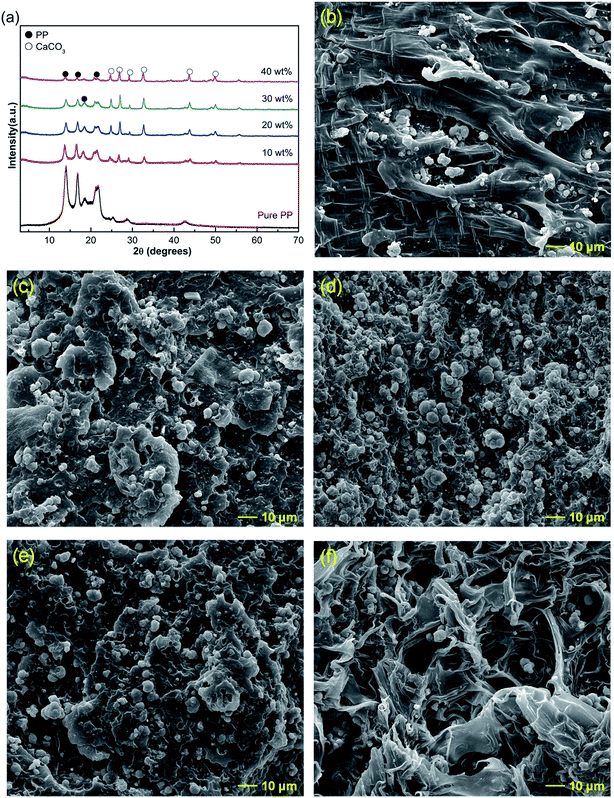 |
| | Fig. 2 (a) XRD patterns, SEM images of PP/modified CaCO3 composites with different percentages of modified CaCO3 ((b) 10%, (c) 20%, (d) 30%, (e) 40%), and (f) the fractured surface of the corresponding sample (e). | |
To illustrate intuitively the feasibility of carbonation product (CaCO3) for applications in plastics, we compared the physical properties of PP/commercial CaCO3 composites, PP/CaCO3 composites, PP/modified commercial CaCO3 composites and PP/modified CaCO3 composites. The variation of the physical properties of PP/CaCO3 composites as a function of CaCO3 and surface modification is shown in Fig. 3, and the impact strength and tensile strength decreased with increasing modified CaCO3 content (Fig. 3a and b). However, the addition of modified CaCO3 to PP could enhance the flexural strength and soften temperature of the composites from 28 MPa and 63 °C for PP to 39.3 MPa and 92.6 °C for PP/modified CaCO3 composites (Fig. 3c and d). While at the addition of 40% CaCO3, the flexural strength and softening temperature of PP/CaCO3 composites could increase up to 34.3 MPa and 119.6 °C. Compared to the PP/CaCO3 composites with the same conditions, the mechanical properties of PP/modified CaCO3 composites to some degrees increased, and the soften temperature accordingly decreased. The results further demonstrated that the morphology of CaCO3 had an obvious effect on the strength of PP-composites. The physical properties of PP-composites filled with commercial CaCO3 and surface modification are also shown in Fig. 3. Compared to CaCO3 and surface modification, they had the same influence on the PP materials. The physical properties of PP/CaCO3 were close to that of PP/commercial CaCO3 (Fig. 3). Therefore, the wollastonite carbonation product (CaCO3) can basically replace commercial CaCO3 used in the plastics field.
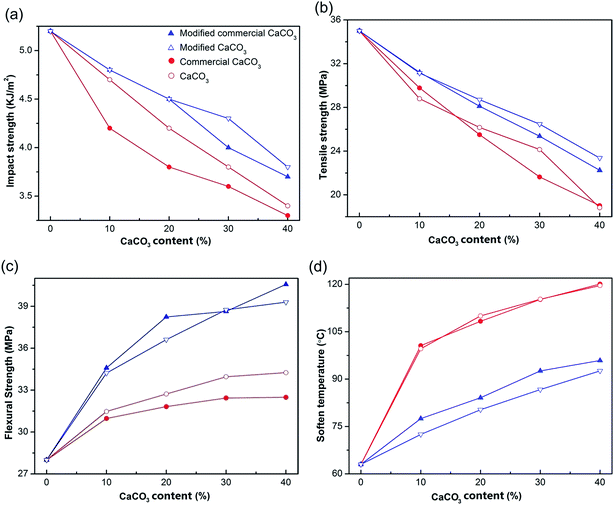 |
| | Fig. 3 Physical properties of PP/CaCO3 composites with different CaCO3 content (10–40%): (a) impact strength, (b) tensile strength, (c) flexural strength and (d) softening temperature. | |
3.3. Polymorph and morphology control of carbonation product
Precipitated calcium carbonate, an important industrial chemical, has been utilized widely for various types of functional application fields such as paper-making, rubber, food, coating, pharmaceuticals and cosmetic manufacturing.38,39 It is generally accepted that the crystal type, morphology and their properties are closely related, and different forms of particles sometimes gave different particle properties even for the same substance.40–42 Therefore, it was very important to design and synthesize CaCO3 with well-controlled polymorph and morphology. In the wollastonite carbonation process, we simultaneously obtained the carbonation product (CaCO3) with a single calcite structure or a single vaterite structure by controlling the reaction temperature or ammonia dosage (Fig. 4 and 5). The carbonation product (CaCO3) with single crystal phase or morphology could potentially be applied to much wider fields.
 |
| | Fig. 4 Polymorph and morphology-controllable preparation of the carbonation product with different temperatures: (a) influence of temperature on the XRD patterns of the carbonation product, (b) influence of temperature on the composition of carbonation products: calcite and vaterite, influence of temperature on the SEM images of carbonation product: (c) 20 °C, (d) 25 °C, (e) 30 °C, (f) 40 °C, (g) 50 °C and (h) 60 °C. | |
 |
| | Fig. 5 Polymorph and morphology controllable preparation of the carbonation product with different ammonia dosages: (a) influence of the ammonia dosage on the XRD patterns of the carbonation product, (b) influence of the ammonia dosage on the composition of carbonation products: calcite and vaterite, influence of the ammonia dosage on the SEM images of the carbonation product: (c) 5 mL, (d) 8 mL, (e) 10 mL and (f) 20 mL. | |
Pure CaCO3, as the product of wollastonite carbonation for CO2 sequestration, was successfully obtained under the action of hydrochloric acid and ammonia through the abovementioned research. Herein, the process was further investigated to elucidate CaCO3 polymorphism as a function of the reaction temperature (Fig. 4) and ammonia dosage (Fig. 5). Fig. 4 revealed the influence of temperature on the structure and morphology of the products. Pure spherical aragonite was detected at 20 °C (Fig. 4a and c). The product composed of spherical vaterite and calcite with a cube shape was synthesized at 25–50 °C (Fig. 4a and d–g). With an increase in temperature from 25 °C to 50 °C, the molar content of vaterite decreased from 90.8% to 62% and that of calcite increased from 9.2% to 38% (ref. 43) (Fig. 4b). Furthermore, pure calcite could be obtained at 60 °C (Fig. 4a), which showed that the increased temperature led to an increase in calcite and a decrease in vaterite. The FT-IR spectral analysis of the products formed at different temperatures were also performed. Fig. S6a† shows the absorption peaks at 877 cm−1 and 744 cm−1 for the ν2 and ν4 vibrations of CO32− in vaterite,44 respectively, and the peaks at 848 cm−1 and 714 cm−1 that are characteristic absorption peaks of calcite45 are consistent with XRD analysis. Fig. 4c–h show the SEM images of the carbonation product at different temperatures.
The influence of the ammonia dosage on the morphology and XRD patterns of the products is also discussed in Fig. 5. Calcite, which was the only crystal phase when using a stoichiometric amount of ammonia, was replaced by vaterite upon the addition of excess ammonia (Fig. 5a). The molar content of calcite with different ammonia dosages was 100%, 26.75%, 0 and 0, respectively, whereas the molar content of vaterite was 0, 73.25%, 100% and 100%, respectively (Fig. 5b). The result was consistent with the previous study.46 The results of the morphology and FT-IR spectra analysis of calcite and vaterite produced with different ammonia dosages (Fig. 5c–f and S6b†) were consistent with the corresponding results under different temperatures.
3.4. Mechanism of wollastonite carbonation
The aqueous carbonation of materials with low porosity such as wollastonite is an irreversible and heterogeneous gas–liquid–solid reaction. In the gas–liquid–solid system, the overall reactions for the mineral carbonation of wollastonite through the proposed process are expressed by eqn (1)–(5). To discuss the reaction mechanism of wollastonite carbonation for CO2 sequestration following the HCl–NH4OH system, the thermodynamic parameters at the standard state in the carbonation process of wollastonite were calculated.47| | |
CaSiO3(s) + 2HCl(aq) → CaCl2(aq) + H2O(l) + SiO2(s), ΔG (kJ mol−1) = −201, K = 1.71 × 1035
| (1) |
| | |
CaCl2(aq) + 2NH4OH(aq) → Ca(OH)2(s) + 2NH4Cl(aq), ΔG (kJ mol−1) = −302, K = 8.66 × 1052
| (2) |
| | |
Ca(OH)2(s) + CO2(g) → CaCO3(s) + H2O(l), ΔG (kJ mol−1) = −122, K = 2.43 × 1021
| (3) |
| | |
2NH4OH(aq) + CO2(aq) → (NH4)2CO3(aq) + H2O(l), ΔG (kJ mol−1) = −276, K = 2.40 × 1048
| (4) |
| | |
CaCl2(aq) + (NH4)2CO3(aq) → CaCO3(s) + 2NH4Cl(aq), ΔG (kJ mol−1) = 107, K = 1.75 × 10−19
| (5) |
From the thermodynamic calculation results, the Gibbs free energy changes (ΔG) are −201, −302, −122 and −276 kJ mol−1 for eqn (1)–(4). They are all less than zero. Their corresponding equilibrium constants (K) are 1.71 × 1035, 8.66 × 1052, 2.43 × 1021, and 2.40 × 1048. They are all greater than zero. Therefore, eqn (1)–(4) can proceed spontaneously at the standard state and regarded as an irreversible exothermic reaction. However, the Gibbs free energy change (ΔG) is 107 kJ mol−1 for eqn (5), more than zero. Its corresponding equilibrium constant (K) is 1.75 × 10−19, close to zero. Eqn (5) cannot proceed spontaneously at the standard state. Compared to the equilibrium constant for eqn (4) (K = 2.40 × 1048), the equilibrium constant for eqn (2) (K = 8.66 × 1052) is greater, which indicates that ammonia was prone to reaction with calcium chloride to from calcium hydroxide. In the desulfurization residue36 (or desulfurization gypsum48) carbonation process, the role of ammonia was to form ammonium carbonate (or ammonium bicarbonate). The reaction mechanism was different in the two systems. Through the abovementioned analysis, the probable mechanism of wollastonite carbonation could be summarized in the following three steps: (a) leaching of Ca2+ from pristine wollastonite, (b) calcium chloride transformed into calcium hydroxide under the action of ammonia, and (c) CO2(g) dissolved in water and reacted with calcium hydroxide to form calcium carbonate.
3.5. Cost estimation of the entire process for wollastonite carbonation
The abovementioned research clearly demonstrated the feasibility of this route proposed in the paper using wollastonite for CO2 sequestration. The entire process consisted of four steps (Fig. 6): (a) Ca2+(aq) leached from the natural wollastonite under the action of hydrochloric acid, (b) CO2(g) dissolved in water to generate CO32−(aq), (c) carbonation product precipitated from the solution under the influence of ammonia, and (d) carbonation product was applied in plastics as a filler. Approximately 1 ton of wollastonite can absorb 350 kg of CO2, where the process requires 0.6 ton of hydrochloric acid, 0.9 ton of ammonia and 3.2 ton of water. In addition, 1 ton wollastonite was also able to produce 510 kg SiO2, 840 kg NH4Cl, and 797 kg CaCO3. These products can also produce a certain economic value. From previous research,28,36,49 we estimated the total cost of the entire wollastonite carbonation process to be around $120, including energy consumption, the value of the by-products and carbonation product (Table 2).
 |
| | Fig. 6 Proposed schematic of the wollastonite carbonation for CO2 sequestration. | |
Table 2 Price of raw materials and treatment energy consumption of the proposed methodsa
| Feed material/products |
Costs ($ per ton) |
Treatment methodology |
Treatment energy consumption (kW h per ton) |
Total costs ($) |
| Note: all the cost of the feed material and products were determined by market price. |
| Wollastonite |
133 |
Heat treatment |
300 |
120 |
| Water |
0.7 |
| HCl |
23 |
Stirring (400 rpm) |
50 |
| NH4OH |
92 |
| SiO2 |
75 |
Filtering |
100 |
| NH4Cl |
85 |
| CaCO3 |
92 |
Drying |
50 |
4. Conclusions
The mineral carbonation of wollastonite for CO2 sequestration under the action of hydrochloric acid and ammonia was studied through experiments and thermodynamic calculations. The ammonia dosage and volume of the Ca2+ leaching solution were the most important process variables influencing the carbonation process. The influence of either the CO2 flow rate or temperature was not as notable as the ammonia dosage and volume of the Ca2+ leaching solution. The carbonation ratio can reach about 93%, and 1 ton of wollastonite was able to sequester 350 kg of CO2. The main crystalline phase of the carbonation product was spherical vaterite, which could meet the relevant standards (HG/T 2226-2010), and their physical properties of PP/CaCO3 composites were very close. The cost of wollastonite carbonation could be reduced by recovering the carbonation byproducts and making full use of carbonation product (CaCO3) with a single calcite structure or a single vaterite structure prepared by controlling the reaction temperature or ammonia dosage during the carbonation process. This mineral carbonation strategy could have interesting potential in CO2 sequestration.
Acknowledgements
This study was supported by the National Science Fund for Distinguished Young Scholars (51225403), the National Natural Science Foundation of China (41572036), the State Key Lab of Powder Metallurgy, Central South University (2015-19), the Hunan Provincial Science and Technology Project (2015TP1006) and the Hunan Provincial Co-Innovation Centre for Clean and Efficient Utilization of Strategic Metal Mineral Resources (2014-405).
Notes and references
- A. Azdarpour, M. Asadullah, E. Mohammadian, R. Junin, H. Hamidi, M. Manan and A. R. M. Daud, Chem. Eng. J., 2015, 264, 425–436 CrossRef CAS.
- A. Sanna, M. Dri, M. R. Hall and M. Maroto-Valer, Appl. Energy, 2012, 99, 545–554 CrossRef CAS.
- P. Friedlingstein, R. M. Andrew, J. Rogelj, G. P. Peters, J. G. Canadell, R. Knutti, G. Luderer, M. R. Raupach, M. Schaeffer, D. P. van Vuuren and C. Le Quéré, Nat. Geosci., 2014, 7, 709–715 CrossRef CAS.
- S. Liu and H. Yang, Energy Technol., 2015, 3, 77–83 CrossRef CAS.
- J. Jin, L. Fu, H. Yang and J. Ouyang, Sci. Rep., 2015, 5, 12429–12437 CrossRef PubMed.
- P. Viebahn, D. Vallentin and S. Höller, Appl. Energy, 2015, 157, 229–244 CrossRef CAS.
- A. Azdarpour, M. Asadullah, E. Mohammadian, H. Hamidi, R. Junin and M. A. Karaei, Chem. Eng. J., 2015, 279, 615–630 CrossRef CAS.
- M. Dri, A. Sanna and M. M. Maroto-Valer, Appl. Energy, 2014, 113, 515–523 CrossRef CAS.
- K. Peng, H. Yang and J. Ouyang, Powder Technol., 2015, 286, 678–683 CrossRef CAS.
- K. Peng, J. Zhang, H. Yang and J. Ouyang, RSC Adv., 2015, 5, 66134–66140 RSC.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 CAS.
- S. Eloneva, A. Said, C. J. Fogelholm and R. Zevenhoven, Appl. Energy, 2012, 90, 329–334 CrossRef CAS.
- L. C. Pasquier, G. Mercier, J. F. Blais, E. Cecchi and S. Kentish, Environ. Sci. Technol., 2014, 48, 5163–5170 CrossRef CAS PubMed.
- J. A. Surface, P. Skemer, S. E. Hayes and M. S. Conradi, Environ. Sci. Technol., 2013, 47, 119–125 CrossRef CAS PubMed.
- M. Niu, X. Li, J. Ouyang and H. Yang, RSC Adv., 2016, 6, 44106–44112 RSC.
- G. Costa, A. Polettini, R. Pomi and A. Stramazzo, J. Hazard. Mater., 2016, 302, 415–425 CrossRef CAS PubMed.
- V. Morales-Flórez, A. Santos, I. Romero-Hermida and L. Esquivias, Chem. Eng. J., 2015, 265, 194–200 CrossRef.
- A. González, N. Moreno and R. Navia, Chemosphere, 2014, 117, 139–143 CrossRef PubMed.
- D. E. Giammar, F. Wang, B. Guo, J. A. Surface, C. A. Peters, M. S. Conradi and S. E. Hayes, Environ. Sci. Technol., 2014, 48, 14344–14351 CrossRef CAS PubMed.
- H. Yan, J. Y. Zhang, Y. C. Zhao, R. Liu and C. G. Zheng, J. Chem. Eng. Jpn., 2015, 48, 937–946 CrossRef CAS.
- C. Julcour, F. Bourgeois, B. Bonfils, I. Benhamed, F. Guyot, F. Bodénan, C. Petiot and É. C. Gaucher, Chem. Eng. J., 2015, 262, 716–726 CrossRef CAS.
- K. Vance, G. Falzone, I. Pignatelli, M. Bauchy, M. Balonis and G. Sant, Ind. Eng. Chem. Res., 2015, 54, 8908–8918 CrossRef CAS.
- K. J. Fricker and A. H. A. Park, Ind. Eng. Chem. Res., 2014, 53, 18170–18179 CrossRef CAS.
- X. L. Wang, A. Sanna, M. M. Maroto-Valer and T. Paulson, Greenhouse Gases: Sci. Technol., 2015, 5, 389–402 CrossRef CAS.
- W. Ding, L. Fu, J. Ouyang and H. Yang, Phys. Chem. Miner., 2014, 41, 489–496 CrossRef CAS.
- D. Daval, I. Martinez, J. Corvisier, N. Findling, B. Goffé and F. Guyot, Chem. Geol., 2009, 265, 63–78 CrossRef CAS.
- M. Kakizawa, A. Yamasaki and Y. Yanagisawa, Energy, 2001, 26, 341–354 CrossRef CAS.
- S. J. Gerdemann, W. K. O'Connor, D. C. Dahlin, L. R. Penner and H. Rush, Environ. Sci. Technol., 2007, 41, 2587–2593 CrossRef CAS PubMed.
- D. Daval, I. Martinez, J. M. Guigner, R. Hellmann, J. Corvisier, N. Findling, C. Dominici, B. Goffé and F. Guyot, Am. Mineral., 2009, 94, 1707–1726 CrossRef CAS.
- E. Ruiz-Agudo, C. V. Putnis, C. Rodriguez-Navarro and A. Putnis, Geology, 2012, 40, 947–950 CrossRef CAS.
- H. J. Zhao, Y. J. Park, D. H. Lee and A. H. A. Park, Phys. Chem. Chem. Phys., 2013, 15, 15185–15192 RSC.
- M. Ghoorah, B. Z. Dlugogorski, R. D. Balucan and E. M. Kennedy, Fuel, 2014, 122, 277–286 CrossRef CAS.
- H. Yang, A. Tang, J. Ouyang, M. Li and S. Mann, J. Phys. Chem. B, 2010, 114, 2390–2398 CrossRef CAS PubMed.
- K. Peng, L. Fu, J. Ouyang and H. Yang, Adv. Funct. Mater., 2016, 26, 2666–2675 CrossRef CAS.
- W. Ding, J. Ouyang and H. Yang, Powder Technol., 2016, 292, 169–175 CrossRef CAS.
- W. Ding, H. Yang and J. Ouyang, RSC Adv., 2015, 5, 67184–67194 RSC.
- J. Feng and M. Chen, Polym. Int., 2003, 52, 42–45 CrossRef CAS.
- G. Hadiko, Y. S. Han, M. Fuji and M. Takahashi, Mater. Lett., 2005, 59, 2519–2522 CrossRef CAS.
- K. Gorna, M. Hund, M. Vučak, F. Gröhn and G. Wegner, Mater. Sci. Eng., A, 2008, 477, 217–225 CrossRef.
- K. Mitsuhashi, N. Tagami, K. Tanabe, T. Ohkubo, H. Sakai, M. Koishi and M. Abe, Langmuir, 2005, 21, 3659–3663 CrossRef CAS PubMed.
- J. Ouyang, H. Yang and A. Tang, Mater. Des., 2016, 92, 261–267 CrossRef CAS.
- X. Li, J. Ouyang, Y. Zhou and H. Yang, Sci. Rep., 2015, 5, 13763–13774 CrossRef PubMed.
- Y. Shen, A. Xie, Z. Chen, W. Xu, H. Yao, S. Li, L. Huang, Z. Wu and X. Kong, Mater. Sci. Eng., A, 2007, 443, 95–100 CrossRef.
- S. Kirboga and M. Öner, CrystEngComm, 2013, 15, 3678–3686 RSC.
- J. Chen and L. Xiang, Powder Technol., 2009, 189, 64–69 CrossRef CAS.
- K. Song, W. Kim, J. H. Bang, S. Park and C. W. Jeon, Mater. Des., 2015, 83, 308–313 CrossRef CAS.
- M. Uibu, K. Tamm, O. Velts-Jänes, P. Kallaste, R. Kuusik and J. Kallas, Fuel Process. Technol., 2015, 140, 156–164 CrossRef CAS.
- M. G. Lee, Y. N. Jang, K. W. Ryu, W. Kim and J. H. Bang, Energy, 2012, 47, 370–377 CrossRef CAS.
- W. J. J. Huijgen, R. N. J. Comans and G.-J. Witkamp, Energy Convers. Manage., 2007, 48, 1923–1935 CrossRef CAS.
- W. J. J. Huijgen, G.-J. Witkamp and R. N. J. Comans, Chem. Eng. Sci., 2006, 61, 4242–4251 CrossRef CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed description of the experimental setup, analytical techniques, and additional results. See DOI: 10.1039/c6ra13908f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 





