
Pressure-induced formation of hydrogen bonds in KNH2 studied by first principles
Abstract
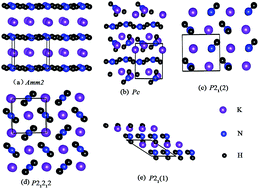
Using particle swarm optimization technique implemented in the CALYPSO code, we have performed systematic research for the structures of KNH2 at pressures up to 20 GPa. Here, phase transition from the ground-state α-KNH2 (monoclinic P21/m) to β-KNH2 (monoclinic P21) occurs at 4 GPa; then, with pressure increasing up to 6.8 GPa, β-KNH2 transforms into γ-KNH2 (monoclinic Pc). By analyzing the partial density of states and charge density, ionic bonding nature between K+ and [NH2]− is revealed and there exists a strong covalent bonding between N and H atoms in NH2 groups in the three structures. Moreover, N–H⋯N hydrogen bonding between neighboring NH2 groups is suggested in β- and γ-KNH2 by investigating the structural details and charge density, which could be favorable for accelerating dehydrogenation behavior of complex metal hydrides.
 Please wait while we load your content...
Please wait while we load your content...

