
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A micropatterning approach to study the influence of actin cytoskeletal organization on polystyrene nanoparticle uptake by BeWo cells†
C.
Muoth
a,
M.
Rottmar
b,
A.
Schipanski
b,
C.
Gmuender
a,
K.
Maniura-Weber
b,
P.
Wick
a and
T.
Buerki-Thurnherr
*a
aParticles-Biology Interactions, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: tina.buerki@empa.ch
bBiointerfaces, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland
First published on 19th July 2016
Abstract
To ensure the safe design and effective application of nanomedical therapies, it is of major importance to understand nanoparticle (NP) uptake mechanisms. Actin-dependent endocytosis has been proposed as a major uptake mechanism for numerous NPs in different cells. However, it is not clear which aspect of the structure and dynamics of the actin cytoskeleton is of relevance in this process. Despite the fact that the cytoskeletal organization is very much dependent on the cellular microenvironment, most mechanistic uptake studies have been performed under classic 2D cell culture conditions, which fail to represent the morphological and mechanical constraints present in biological tissues. In this study, human choriocarcinoma BeWo cells were physically constrained on micropatterns of different geometries to study the uptake of 80 nm, 240 nm and carboxylate 300 nm polystyrene particles and the dependence on different actin structures. Interestingly, major differences in the actin cytoskeletal organization of BeWo cells did not affect the intracellular distribution and amount of internalized polystyrene NPs for all investigated particle sizes and modifications. Therefore, we suggest a more central role for actin dynamics in actin-dependent NP endocytosis, rather than the overall actin organization.
Introduction
The concept of nanomedicine is based on the unique physico-chemical properties of nanoparticles (NPs) that allow a broad range of applications in different medical fields such as imaging (e.g. semiconductor, metal or metal oxide NPs),1 drug delivery (e.g. polymeric NPs),2 organ repair (e.g. silica or iron oxide NPs)3 or photodynamic therapies (e.g. inorganic NPs with tunable optical properties).4 Many of these applications require NP uptake by the target tissue for optimal efficiency and to reduce side effects to a low level. However, despite the presence of many NP uptake studies, the current knowledge on NP internalization in complex biological tissues is still limited. It has emerged that the uptake and the intracellular fate of NPs are greatly dependent on the tissue type, with their respective delivery routes and the physico-chemical properties of the NPs (i.e. size, shape, material, surface charge and functionalization; for extensive reviews see ref. 1 and 5–10). In addition, many studies have reported the presence of NPs in membrane-bound vesicles and/or reduced uptake at low temperatures, both of which are indicative of an active endocytosis uptake mechanism (reviewed in ref. 11–14).The actin cytoskeleton plays a fundamental role in different cellular processes, including many types of endocytosis, such as phagocytosis, macropinocytosis, circular dorsal ruffles or clathrin-mediated endocytosis (CME).15,16 Actin assembly can create protrusions that engulf extracellular materials. Actin also supports the invagination of membrane segments into the cytoplasm, scission of new vesicles from the plasma membrane and intracellular movement of the vesicles. In CME in yeast, vesicle budding is completed by the formation of a tubular invagination that is driven by the attachment of filamentous actin (F-actin), which occurs concomitantly with nucleation of actin at the plasma membrane.17,18 Similar events have been shown for mammalian cells, where a close association between F-actin accumulation and vesicle formation was observed at a late stage of CME.19,20 In addition, there is also evidence that actin not only plays a mechanical role, but also a structural role in CME, where it guides the endocytic machinery to a specific site on the plasma membrane (reviewed in ref. 21). A central role for actin dynamics in NP uptake by different cell types has been proposed in several studies employing pharmacological inhibitors that affect actin reorganization and G/F-actin ratios, such as cytochalasin or latrunculin.22–25 Although there is some indication that actin-dependent internalization may be dependent on particle size and aspect ratio/geometry,26,27 the use of different cell types, pharmacological inhibitors or NP types makes it difficult to compare different studies and to obtain a clear mechanistic insight. As such, treatment of macrophages with cytochalasin B resulted in the reduced uptake of 1000 nm, but not that of 20 nm polystyrene (PS) NPs,25 while cytochalasin D treatment significantly diminished the internalization of small 20 nm silica NPs in A549 lung epithelial cells.24 In addition, the use of such pharmacological inhibitors has some drawbacks, including a low specificity and cytotoxicity at increased concentrations or prolonged exposure times. Furthermore, cytochalasin can also affect other biological processes unrelated to actin polymerization, such as protein synthesis28 or glucose transport.29 Besides actin dynamics, we hypothesize that the global organization of actin filaments, as well as their assembly into higher-order structures, such as contractile bundles or three-dimensional networks may be implicated in cellular NP uptake and subcellular localization.
The microcontact printing (μCP) technique presents an interesting approach to mechanically controlling the organization of the actin cytoskeleton and when creating defined patterns, a more tissue-like microenvironment can be mimicked.30 On substrates produced by μCP, single cells can be cultivated on cell-adhesive islands of defined geometry and size to manipulate and control the cell spreading area, shape, cytoskeletal organization and cell contractility, among other factors.31–34 It has been previously shown that geometric cues can guide the formation of actin stress fibers and lamellipodia.35 While cell adhesive areas with concave edges promote the formation of actin stress fibers,34 those with convex edges are strong cues for the formation of lamellipodia.36 Both actin stress fibers and lamellipodia have been shown to influence cell polarity.36,37 Micropatterns with increased substrate stiffness and shapes that promote high cytoskeletal tension enhanced the osteogenesis of mesenchymal stem cells,35,38,39 whereas unspread, round cells tend to express elevated markers of adipogenesis.35,39,40 Taken together, these reports demonstrate the potential of μCP to enable studies on basic cell responses to individual cues with shape-normalized single cells.
In this study, μCP was employed to investigate the influence of different actin cytoskeletal organizations on uptake and internalization of PS particles by BeWo cells. Therefore, BeWo cells were cultivated on four different pattern geometries, resulting in the same cell surface area but different shapes, which induced highly distinct organization of the actin cytoskeleton. Nontoxic, fluorescent and monodisperse PS particles were used as model particles for the uptake studies. BeWo cells were exposed to 80 nm, 240 nm and carboxylate 300 nm PS particles, followed by quantitative uptake analysis using confocal microscopy and a digital image analysis method. We achieved the successful normalization of BeWo cell shape and distinct actin filament arrangements, a prerequisite for subsequent studies on actin-dependent NP uptake. Intracellular distribution and the amount of internalized NPs by shape-normalized BeWo cells were independent of the cytoskeletal organization for all investigated sizes and modifications of PS particles. This model presents a novel approach to studying the role of actin structures in NP uptake, kinetics and intracellular fate over time, and its dependency on different NP properties and cell types. A detailed knowledge of these NP-cell interactions is crucial for the development of targeted drug delivery systems.
Results and discussion
Particle characterization and cytotoxicity assessment
Commercially available, fluorescence labelled PS particles were selected as model NPs because actin-dependent endocytosis has been observed for this type of NP.22,25,41 PS particles with different physico-chemical properties were included to further investigate whether NP uptake by micropatterned cells is size – (plain 80 nm and 240 nm PS NPs) or charge-dependent (plain 240 nm particles and 300 nm COOH–PS NPs). This size range was chosen because 300 nm is considered to be the relevant cut-off size for nanomedical particles, where unique medical effects are often observed only for particles larger than 100 μm.42 PS particles were characterized in terms of primary particle size, hydrodynamic particle size, zeta potential and stability of the fluorescent dye. Results for the selected particles from previous work,43,44 as well as new characterization data from this study, are summarized in Table 1.| Plain | COOH | ||
|---|---|---|---|
| 80 nm | 240 nm | 300 nm | |
| a Abbreviations: DD double distilled; TEM transmission electron microscopy. b According to manufacturer's information. c Grafmueller et al.44 d Grafmueller et al.43 e Experimentally determined (mean ± SD). | |||
| Diameter (nm) | 87b | 240 | 302.7d |
| Diameter TEM (nm) | 78.1 ± 20.5c | 220.5 ± 5.1d | 289.4 ± 10.2d |
| Hydrodynamic diameter (nm) in DD water | 97 ± 0.8c | 230 ± 65.3d | 283 ± 85.2d |
| Hydrodynamic diameter (nm) in growth mediume | 162 ± 5.3 | 207 ± 10.9 | 312 ± 9.4 |
| Initial no. of particles per mL in growth medium | 2.18 × 1012 | 1.70 × 109 | 7.50 × 108 |
| Particle surface (nm2) per mL in growth medium | 1.31 × 1016 | 2.59 × 1014 | 1.97 × 1014 |
| Zeta potential in 10 mM NaCl (mV) | −50 ± 9.7c | −20.5 ± 2.7d | −55.6 ± 6.1d |
| Zeta potential in growth medium (mV)e | −10.9 ± 0.8 | −10.9 ± 0.6 | −9.3 ± 0.97 |
| Fluorescent dyeb | Yellow green | Yellow | Yellow green |
| Excitation/emissionb | 485/528 | 485/520 | 485/528 |
| Manufacturer | Polyscience Inc | Spherotec Inc | Polyscience Inc |
TEM analysis was performed in a previous study and showed a primary particle size of 78.1 ± 20.5 nm for the plain 80 nm PS particles,44 220.5 ± 5.1 nm for the plain 240 nm PS particles43 and 289.4 ± 10.2 nm for the 300 nm COOH–PS particles.43 In suspension, hydrodynamic diameters of 80 nm, 240 nm and 300 nm COOH–PS particles increased to 97 ± 0.8 nm, 230 ± 65.3 nm and 283 ± 85.2 nm in water and to 162 ± 5.3 nm, 207 ± 10.9 nm and 312 ± 9.4 nm in growth medium, respectively (ESI Fig. 1†). The zeta potential was negative for all PS particles suspended in 10 mM sodium chloride, but for 80 nm and 300 nm COOH–PS particles, it was significantly lower than for 240 nm PS particles. Since 240 nm and 300 nm PS particles were similar in size but with different zeta potential, they were included to investigate whether NP surface charges affect their internalization by micropatterned cells.
When working with fluorescent NPs, a common problem is the release of labile dye in a biological milieu, as free fluorophores may significantly alter the in vitro and in vivo outcomes.45–48 Therefore, we assessed the stability of the fluorescent dye in growth medium at 37 °C over 72 h, and no decrease in fluorescence intensity or any leakage of free dye from the different PS particles used in this study were observed (ESI Fig. 2A and B†).
An important prerequisite to study the dependency of NP uptake on cytoskeletal organization is the absence of cytotoxic effects. Using the MTS assay, we confirmed that none of the PS particles decreased the viability of BeWo cells within 24 h of exposure (ESI Fig. 3A and B†).
Distinct cytoskeletal organization through micropatterning
In a μCP approach, we explored whether different cytoskeletal organization influenced NP uptake by single cells. The human epithelial choriocarcinoma cell line BeWo was chosen, as it expresses a wide range of different transporters, including various exporters for the efflux of toxic xenobiotics.49 Furthermore, uptake and translocation of different NPs have been shown by BeWo cells in vitro,50–52 and the syncytiothrophoblast ex vivo.43,53–55 The possibility to induce major differences in intracellular organization in a purely mechanical way, without the need for pharmacological inhibitors renders μCP a very interesting tool for mechanistic uptake studies. An additional advantage of the μCP technique in particular for NP uptake studies is that significantly prolonged exposure times (24 h or more, depending on the cell division rate) can be studied, as opposed to only few hours for the use of actin inhibitors.56 Moreover, it has been shown for certain cell types such as endothelial cells, that the composition and stiffness of micropatterned substrates influence cell architecture, mechanics and the cytoskeletal organization in a similar way to that found for cells in situ within organs or tissues.57–59In this study, BeWo cells were grown on adhesive micropatterns of different geometries (disc, H, crossbow (CB) and Y) (Fig. 1A) in order to achieve differences in cytoskeletal organization without changing the cell surface area. The optimal pattern size, which best supported the spreading of BeWo cells was 1600 μm2. On smaller micropatterns (1100 and 700 μm2), cells displayed a roundish morphology while on larger patterns (>2000 μm2), they were not able to span over the entire adhesive area (data not shown). Major differences in the actin cytoskeleton occurred between Y patterns with three non-adhesive areas, CB and H patterns with two non-adhesive areas and disc patterns lacking non-adhesive areas. On Y patterns, filamentous actin (F-actin) stress fibers were concentrated along the whole outline of the cell, whereas on CB and H patterns, they only formed along two borders of the cell. In contrast, BeWo cells spread on the disc patterns displayed a strong actin network in the center of the cell where the cytoskeleton spanned over the nucleus (Fig. 1B). Furthermore, the formation of lamellipodial extensions and filopodial protrusions were detectable on adhesive areas in BeWo cells grown on disc, H and CB patterns (Fig. 2). To confirm that the cytoskeletal organization was reproducible, actin heatmaps of 20 individual cells per pattern were made (Fig. 1C). Actin heatmaps showed a highly reproducible and distinct arrangement of the actin cytoskeleton within each pattern demonstrating the successful normalization of BeWo cells.
 | ||
| Fig. 1 Normalization of BeWo cells on disc, H, CB and Y micropatterns. The four different patterns on a chip without (A) and with BeWo cells (B). The actin cytoskeleton was visualized by phalloidin (red), cell nuclei by DAPI (blue) and the adhesive patterns by a fluorescent fibronectin coating (gray). Differential organization of the actin cytoskeleton on the different patterns was evident (63× magnification). Heat maps were calculated from 20 overlaid z-stack images per pattern geometry, showing the distribution of actin stress fibers (C). Red indicates a high pixel intensity of F-actin, whereas blue represents a low intensity. | ||
 | ||
| Fig. 2 Formation of actin protrusions in cell regions on adhesive areas. BeWo cells were spread on disc (A), H (B) and CB (C) patterns and stained for F-actin (red), showing filopodia-like protrusions and lamellipodia-like structures on the adhesive areas on the border of the cells. Actin protrusions of BeWo cells grown on Y patterns (D) were less prominent. | ||
NP uptake by shape-normalized BeWo cells
BeWo cells that were completely spread on the different micropatterns were exposed to PS particles in order to specifically study the potential effects of differences in cytoskeletal organization on the amount and distribution of internalized NPs. Addition of NPs before or during cell spreading would prevent the achievement of the study aim, since unspread cells have a randomly organized actin cytoskeleton and NPs could interfere with cell adhesion and spreading.60 Experiments using different NP concentrations were performed to identify a suitable NP concentration that resulted in a sufficient number of intracellular NPs in the majority of the cells to quantitatively analyze NP uptake by confocal microscopy (data not shown). While a low concentration of 10 μg mL−1 was sufficient for 240 nm (Fig. 3B) and 300 nm COOH–PS particles (Fig. 3C), a higher concentration of 100 μg mL−1 was required for 80 nm PS particles (Fig. 3A). An exposure time of 14 h was chosen because most BeWo cells had not yet entered mitosis, resulting in a high number of single cells that could be included in the analysis.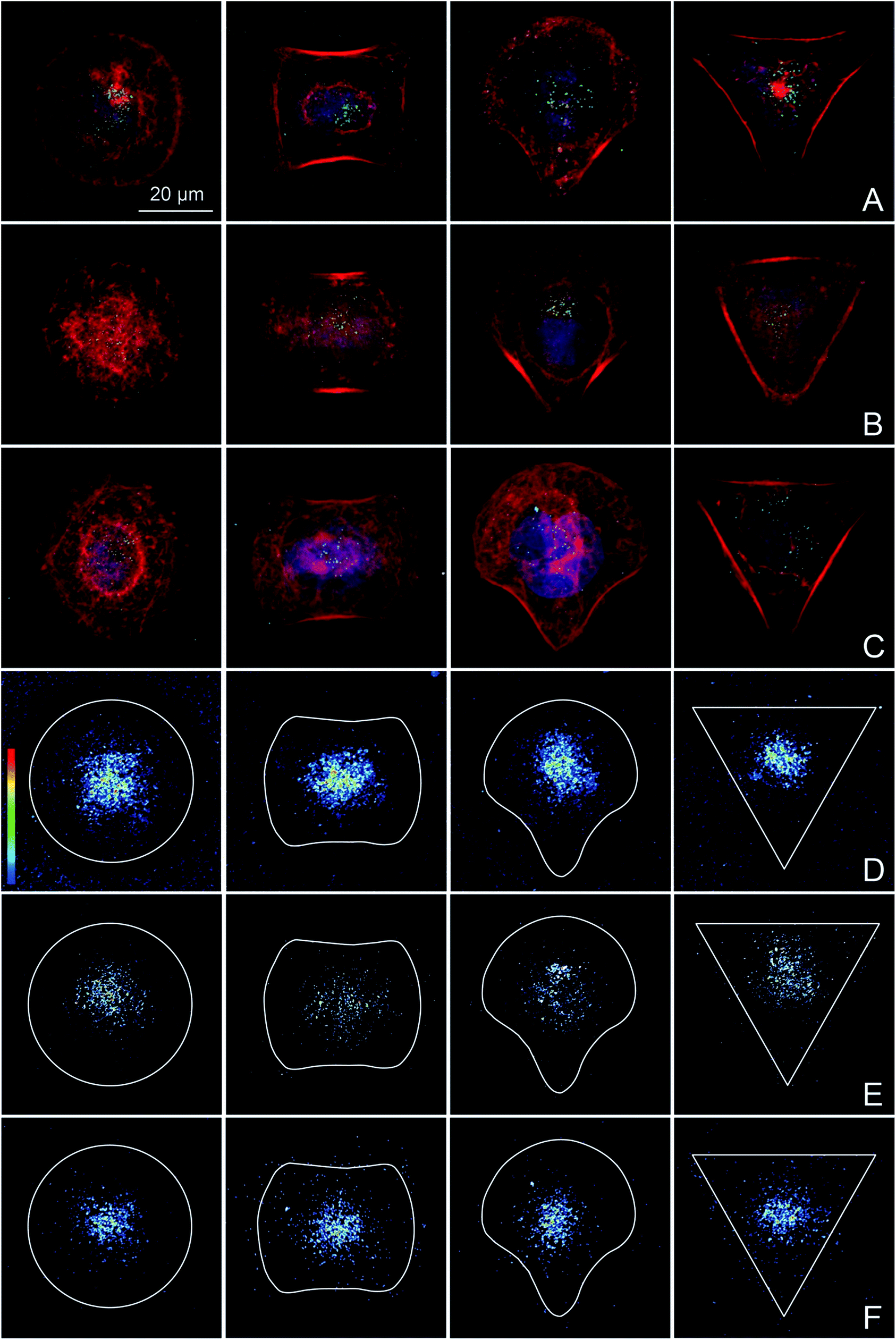 | ||
| Fig. 3 NP uptake by shape-normalized single cells. BeWo cells cultivated on adhesive micropatterns (disc, H, CB and Y) were exposed to 100 μg mL−1 of 80 nm (A) and 10 μg mL−1 of 240 nm (B) and 300 nm green fluorescent COOH–PS particles (C) for 14 h. Cells were stained for F-actin (red) and DAPI (blue). Heatmaps were calculated from 20 overlaid z-stack images per pattern geometry, showing the distribution of internalized 80 nm (D), 240 nm (E) and 300 nm COOH–PS particles (F). Red indicates a high pixel intensity of F-actin, whereas blue represents a low intensity. | ||
Heatmaps were made to show the average distribution of internalized PS particles. After an exposure time of 14 h, uptake was observed for all shape-normalized BeWo cells exposed either to 80 nm (Fig. 3D), 240 nm (Fig. 3E) or 300 nm COOH–PS particles (Fig. 3F). As expected, for single cell analysis,61 the amount of internalized NPs was highly variable between different cells, even if cultivated on the same pattern. Independent of the different pattern geometries, all NPs were located predominantly in the perinuclear region, probably in the form of NP-filled vesicles. Although it may appear from several individual images in Fig. 3 that actin is also very much localized around the nuclei, heatmaps from 20 individual cells clearly showed that a consistent perinuclear localization of actin is only true for cells spread on the disc patterns (Fig. 2). Therefore, perinuclear actin structures are most likely not responsible for the observed nuclear localization of the NPs. Most importantly, NP heatmaps did not show any obvious differences in the amount or intracellular distribution of internalized NPs, despite the presence of highly distinct actin organization on the four pattern geometries.
In addition to the qualitative observations, the quantity of internalized NPs was analyzed by applying a macro specifically designed to reliably measure NP uptake by single cells (“Particle_in_Cell-3D”61 for ImageJ). This macro requires the use of fluorescently labelled NPs, as well as a staining procedure to visualize the cell border or cell membrane. However, direct labelling of the cell membrane (e.g. with cell tracker dyes, DiI, DiO or DiD) led to the partial detachment of the cells from the patterns after fixation (data not shown). Therefore, labelling of the actin cytoskeleton was used as an indirect approach to identify the cell border. Integrated pixel intensities of internalized NPs were calculated for 20 cells per pattern geometry, using the same z-stack images as for the heatmaps (Fig. 4). Comparison of PS particle uptake by BeWo cells grown on disc, H, CB and Y patterns revealed no differences for either the 80 nm (Fig. 4A) or 240 nm PS particles (Fig. 4B). A slightly increased uptake was observed only for 300 nm COOH–PS particles by cells grown on Y patterns, compared to CB and H patterns (Fig. 4C), but this trend was not observed for 80 nm or 240 nm PS particles. As this increase was very modest and not consistent for all NP types, it is unlikely that cells with high levels of peripheral stress fibers incorporate more NPs than cells with a more central F-actin meshwork.
 | ||
| Fig. 4 Quantitative analysis of PS particle uptake by shape-normalized single cells grown on the different patterns. NP uptake of 20 individual cells per pattern was analyzed using the ImageJ macro “Particle_in_Cell-3D”. Tukey box-and-whiskers plots show the integrated NP pixel intensity per cell surface area for all pattern geometries. Quantitative NP uptake by BeWo cells was calculated for (A) 80 nm, (B) 240 nm and (C) 300 nm COOH–PS particles. | ||
Our quantitative analysis indicates that actin cytoskeletal organization is not a key determinant of NP internalization. Specifically, PS NP uptake was independent of the presence of wide, peripheral stress fibers, filopodia or lamellipodial structures (Fig. 1C and 2). In addition, a more peripheral or central localization of most F-actin structures did not have any influence on the amount or intracellular distribution of PS NPs either (Fig. 1C). However, due to cell type-specific differences in NP uptake, it will be important to confirm these findings for other cell types. Moreover, it appears that cell contractility is not implicated in PS NP uptake, since previous work suggested that the level of contractile forces was altered in cells grown on different micropatterns.34,36 According to these studies, cell contractility in micropatterned BeWo cells is expected to be highest on Y patterns forming three convex edges, intermediate on CB and H patterns forming two convex edges and lowest on disc patterns forming no such edges. However, to unambiguously prove that NP internalization is independent of cell contractility, contractile forces of cells on different micropatterns have to be accurately determined by appropriate force measurement methods. Finally, we suggest that the dynamic reorganization of the actin filaments, which was not significantly affected in our μCP study, is of key relevance for the active endocytic NP uptake as observed in previous studies.22–25 This is supported by the fact that the presence of pharmacological inhibitors interfering with polymerization, depolymerization or rearrangement of the actin cytoskeleton resulted in the decreased uptake of several NPs.22–25
The current knowledge on NP uptake mechanisms by different cell types ranges from actin-dependent endocytosis to non-endocytic pathways with no clear correlation to the physico-chemical properties of the particles. For example, dos Santos et al. described an energy dependent uptake process for 40 nm and 200 nm COOH–PS NPs in HeLa and glial astrocytoma 1321N1 cells.22 In contrast, a nonendocytic uptake pathway was suggested for 78 nm and 200 nm PS NPs in pulmonary macrophages.62 A more recent study in primary macrophages and a monocytic cell line has identified endocytosis, macropinocytosis or phagocytosis as the involved uptake mechanisms for 100 nm COOH– and NH2–PS NPs.41 These contradictory results clearly demonstrate the need for more detailed studies on NP uptake mechanisms, including, but not limited to the role of endocytosis and the actin cytoskeleton.
Conclusions
In this study, single BeWo cells with the same projected surface area, but highly distinct organization of the actin cytoskeleton were successfully obtained using commercial micropatterns with different geometries. Using μCP technology, we were able to exclude a major role of the global actin organization in the internalization of PS particles by BeWo cells. We propose that future work on actin-dependent NP uptake mechanisms should focus on the precise role of the dynamic reorganization of actin microfilaments. For such studies, the use of pharmacological inhibitors is inevitable, as major changes in actin dynamics or G/F-actin ratios cannot be easily achieved by a μCP approach. However, to obtain mechanistic insights that are predictive of the in vivo situation in biological tissues, such inhibitor studies should be performed on micropatterned cells or 3D microtissues, rather than highly artificial 2D cell cultures.As nanotechnology is becoming a vital player in medicine, with various biomedical applications, such as drug delivery, therapeutics, diagnosis or imaging, it is necessary to understand the mechanisms by which NPs are internalized by cells. A comprehensive knowledge of NP internalization pathways in relation to distinct physico-chemical NP properties may enable the development of novel strategies to enhance or reduce NP uptake at specific sites.
Experimental section
Micropatterns
Micropatterns coated with fluorescent fibronectin with excitation at 650 nm were acquired from CYTOO Cell Architects (Grenoble, France). A CYTOOchip™ was 20 × 20 mm and contained four different geometries (disc, crossbow (CB), H and Y) (Fig. 1A) in three sizes (700, 1100 and 1600 μm2 per pattern) and a control area uniformly covered with A650-labeled fibronectin.Cell culture
BeWo (ATCC) cells, a human placental choriocarcinoma cell line with cytotrophoblastic characteristics, were cultured in growth medium (Ham's F-12K medium (Gibco, Luzern, Switzerland) supplemented with 10% fetal calf serum (Invitrogen, Basel, Switzerland), 2 mM L-glutamine (Gibco, Luzern, Switzerland) and 1% penicillin/streptomycin (Gibco, Luzern, Switzerland)). Cells were cultivated in a humidified incubator at 37 °C with 5% CO2 atmosphere.Polystyrene particles and characterization
Fluorescently yellow-green-labeled polystyrene (PS) particles of 80 nm (ex/em: 485/528 nm) and carboxylate-modified (COOH) 300 nm (ex/em: 485/528 nm) were purchased from Polyscience (Warrington, USA). Yellow-green-labeled 240 nm PS particles (ex/em: 485/520 nm) were purchased from Spherotech (Lake Forest, USA). Particles were vortexed for 2 min before preparation of experimental dilutions and exposure of cells.The zeta potential of the particles in growth medium was determined with a Zetasizer NanoZS (Malvern Instruments, Malvern, UK).
Particle size distribution in growth medium and double distilled (DD) water was determined by nanoparticle tracking analysis (Nanosight LM 20 System, Amesbury, UK). Prior to analysis, growth medium and DD water were filtered using a 0.02 μm Anotop® 25 syringe filter (Whatman GmbH, Dassel, Germany). Results were normalized to the area under the NP concentration/size curve.
The stability of the fluorescence was assessed by analyzing the loss of fluorescence after incubation of 25 μg mL−1 PS particles diluted in growth medium at 37 °C for 3, 6, 24, 48 and 72 h using a microplate reader (Biotek Synergy, Winooski, USA) with excitation and emission wavelengths of 485 nm and 528 nm. The loss of fluorescence was measured before and after filtration through a 0.1 μm syringe filter at the end of each incubation time.
In vitro cell viability assay
The MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay was used to test in vitro cytotoxicity of the different PS particles based on a colorimetric process to evaluate mitochondrial dehydrogenase activity of BeWo cells. In the presence of phenazine methosulfate, viable BeWo cells reduce MTS reagent to a colored formazan. 24 h prior to treatment, BeWo cells were seeded into a 96-well plate at 8000 cells per well. BeWo cells were exposed to 6.25, 12.5, 25, 50 and 100 μg mL−1 of plain 80 nm and 240 nm PS NPs and 300 nm COOH–PS particles for 3 h and 24 h at 37 °C and 5% CO2. Exposure of cells to 1, 10, 100 and 1000 μM CdSO4 served as positive control, whereas cells without treatment were used as the negative control. After incubation, an MTS assay (CellTiter96® AQueous One Solution Cell Proliferation Assay; Promega, Dübendorf, Switzerland) was performed according to the manufacturer's instructions. Results were presented as mean percentage of the untreated control from three independent experiments.Uptake analysis on micropatterns
CYTOOchips™ were placed into P35 plastic culture dishes at 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 BeWo cells per chip. According to the manufacturer's protocol, dishes were not moved for 30 min after cell seeding to provide a uniform cell distribution and sedimentation, and were then transferred to the incubator. PS particles were added as soon as BeWo cells had spread onto the entire pattern area (after approximately 3 h) at a final concentration of 100 μg mL−1 for 80 nm and 10 μg mL−1 for 240 nm and 300 nm COOH–PS particles. After 14 h cells were fixed with 4% PFA (Sigma-Aldrich, Buchs, Switzerland)/0.2% TX100 (Sigma-Aldrich, Buchs, Switzerland) for 10 min, washed and incubated with Alexa Fluor 546 phalloidin (Invitrogen, Basel, Switzerland, 1:200) and DAPI (4′,6-diamidin-2-phenylindol) (Sigma-Aldrich, Buchs, Switzerland) for 1 h at room temperature. Prior to microscopic analysis CYTOOchips™ were mounted in Mowiol 4-88 (Sigma-Aldrich, Buchs, Switzerland).
000 BeWo cells per chip. According to the manufacturer's protocol, dishes were not moved for 30 min after cell seeding to provide a uniform cell distribution and sedimentation, and were then transferred to the incubator. PS particles were added as soon as BeWo cells had spread onto the entire pattern area (after approximately 3 h) at a final concentration of 100 μg mL−1 for 80 nm and 10 μg mL−1 for 240 nm and 300 nm COOH–PS particles. After 14 h cells were fixed with 4% PFA (Sigma-Aldrich, Buchs, Switzerland)/0.2% TX100 (Sigma-Aldrich, Buchs, Switzerland) for 10 min, washed and incubated with Alexa Fluor 546 phalloidin (Invitrogen, Basel, Switzerland, 1:200) and DAPI (4′,6-diamidin-2-phenylindol) (Sigma-Aldrich, Buchs, Switzerland) for 1 h at room temperature. Prior to microscopic analysis CYTOOchips™ were mounted in Mowiol 4-88 (Sigma-Aldrich, Buchs, Switzerland).
Z-stack images were acquired with a 63× magnification oil immersion objective on a confocal laser scanning microscope (LSM 780, Zeiss, Feldbach, Switzerland) using the identical color intensity threshold for each type of NP. For each of the different experimental conditions 20 individual cells were imaged, which is a similar cell number to that used in other NP uptake studies.63–65 After image processing (ZEN 2011, Zeiss, Feldbach, Switzerland; Photoshop CS6), heatmaps were determined from the 20 overlaid z-stacks/pattern for each pattern geometry, in order to visualize the average distribution of actin and internalized PS particles. Quantification of internalized 80 nm, 240 nm and 300 nm COOH–PS particles by single cells was performed from heatmap z-stack images using the ImageJ macro “Particle_in_Cell-3D”.61 Briefly, images of the actin cytoskeleton were used to recalculate the cell volume in which the particles were quantified. The total fluorescence of the internalized NPs was digitally assessed by the sum of all NP pixel intensities within each cell volume, which is assumed to be proportional to the amount of uptaken NPs.
Statistics
For statistical analysis of NP uptake, Tukey box-and-whiskers plots were created to compare median values of the integrated NP pixel intensities between the pattern geometries (Prism 6, GraphPad, California, USA). Values with a distance larger than 1.5 times the interquartile range from the 25th and 75th percentile were considered as outliers and shown individually. Results were analyzed by one-way ANOVA with Tukey's multiple comparison test and regarded as statistically significant at a p-value below 0.05.Acknowledgements
We would like to thank Adriano A. Torrano for technical support in using the macro “Particles_in_Cell-3D”. This project has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no 309329 (NANOSOLUTIONS).References
- J. Nam, N. Won, J. Bang, H. Jin, J. Park, S. Jung, S. Jung, Y. Park and S. Kim, Adv. Drug Delivery Rev., 2013, 65, 622–648 CrossRef CAS PubMed.
- A. Z. Mirza and F. A. Siddiqui, Int. Nano Lett., 2014, 4, 1–7 CrossRef.
- A. Meddahi-Pelle, A. Legrand, A. Marcellan, L. Louedec, D. Letourneur and L. Leibler, Angew. Chem., 2014, 53, 6369–6373 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- A. Albanese, P. S. Tang and W. C. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- H. Kettiger, A. Schipanski, P. Wick and J. Huwyler, Int. J. Nanomed., 2013, 8, 3255–3269 Search PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 1–11 CrossRef PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- L. Treuel, X. Jiang and G. U. Nienhaus, J. R. Soc., Interface, 2013, 10, 1–14 CrossRef PubMed.
- O. L. Mooren, B. J. Galletta and J. A. Cooper, Annu. Rev. Biochem., 2012, 81, 661–686 CrossRef CAS PubMed.
- M. Kaksonen, C. P. Toret and D. G. Drubin, Nat. Rev. Mol. Cell Biol., 2006, 7, 404–414 CrossRef CAS PubMed.
- F. Z. Idrissi, H. Grotsch, I. M. Fernandez-Golbano, C. Presciatto-Baschong, H. Riezman and M. I. Geli, J. Cell Biol., 2008, 180, 1219–1232 CrossRef CAS PubMed.
- M. Kaksonen, C. P. Toret and D. G. Drubin, Cell, 2005, 123, 305–320 CrossRef CAS PubMed.
- C. J. Merrifield, D. Perrais and D. Zenisek, Cell, 2005, 121, 593–606 CrossRef CAS PubMed.
- D. Yarar, C. M. Waterman-Storer and S. L. Schmid, Mol. Biol. Cell, 2005, 16, 964–975 CrossRef CAS PubMed.
- A. E. Engqvist-Goldstein and D. G. Drubin, Annu. Rev. Cell Dev. Biol., 2003, 19, 287–332 CrossRef CAS PubMed.
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, PLoS One, 2011, 6, e24438 CAS.
- C. P. García, V. Sumbayev, D. Gilliland, I. M. Yasinska, B. F. Gibbs, D. Mehn, L. Calzolai and F. Rossi, Sci. Rep., 2013, 1326, 1–7 Search PubMed.
- J. S. Nowak, D. Mehn, P. Nativo, C. P. Garcia, S. Gioria, I. Ojea-Jimenez, D. Gilliland and F. Rossi, Toxicol. Lett., 2014, 224, 84–92 CrossRef CAS PubMed.
- B. Prietl, C. Meindl, E. Roblegg, T. R. Pieber, G. Lanzer and E. Frohlich, Cell Biol. Toxicol., 2014, 30, 1–16 CrossRef CAS PubMed.
- H. Herd, N. Daum, A. T. Jones, H. Huwer, H. Ghandehari and C. M. Lehr, ACS Nano, 2013, 7, 1961–1973 CrossRef CAS PubMed.
- H. Meng, S. Yang, Z. X. Li, T. Xia, J. Chen, Z. X. Ji, H. Y. Zhang, X. Wang, S. J. Lin, C. Huang, Z. H. Zhou, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4434–4447 CrossRef CAS PubMed.
- D. A. Ornelles, E. G. Fey and S. Penman, Mol. Cell. Biol., 1986, 6, 1650–1662 CrossRef CAS PubMed.
- A. L. Rampal, H. B. Pinkofsky and C. Y. Jung, Biochemistry, 1980, 19, 679–683 CrossRef CAS PubMed.
- M. Thery, J. Cell Sci., 2010, 123, 4201–4213 CrossRef CAS PubMed.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Biotechnol. Prog., 1998, 14, 356–363 CrossRef CAS PubMed.
- K. K. Parker, A. L. Brock, C. Brangwynne, R. J. Mannix, N. Wang, E. Ostuni, N. A. Geisse, J. C. Adams, G. M. Whitesides and D. E. Ingber, FASEB J., 2002, 16, 1195–1204 CrossRef CAS PubMed.
- R. Singhvi, A. Kumar, G. P. Lopez, G. N. Stephanopoulos, D. I. C. Wang, G. M. Whitesides and D. E. Ingber, Science, 1994, 264, 696–698 CAS.
- M. Thery, A. Pepin, E. Dressaire, Y. Chen and M. Bornens, Cell Motil. Cytoskeleton, 2006, 63, 341–355 CrossRef CAS PubMed.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872–4877 CrossRef CAS PubMed.
- J. James, E. D. Goluch, H. Hu, C. Liu and M. Mrksich, Cell Motil. Cytoskeleton, 2008, 65, 841–852 CrossRef PubMed.
- M. Thery, V. Racine, M. Piel, A. Pepin, A. Dimitrov, Y. Chen, J. B. Sibarita and M. Bornens, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19771–19776 CrossRef CAS PubMed.
- J. Lee, A. A. Abdeen, T. H. Huang and K. A. Kilian, J. Mech. Behav. Biomed. Mater., 2014, 38, 209–218 CrossRef CAS PubMed.
- R. McBeath, D. Pirone, C. Nelson, K. Bhadriraju and C. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
- J. Lee, A. A. Abdeen, D. Zhang and K. A. Kilian, Biomaterials, 2013, 34, 8140–8148 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delecher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailander, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, J. Nanomed. Nanotechnol., 2013, 9, 1–14 CrossRef CAS PubMed.
- S. Grafmueller, P. Manser, L. Diener, P. A. Diener, X. Maeder-Althaus, L. Maurizi, W. Jochum, H. F. Krug, T. Buerki-Thurnherr, U. von Mandach and P. Wick, Environ. Health Perspect., 2015 DOI:10.1289/ehp.1409271.
- S. Grafmueller, P. Manser, L. Diener, L. Maurizi, P. A. Diener, H. Hofmann, W. Jochum, H. F. Krug, T. Buerki-Thurnherr, U. von Mandach and P. Wick, Sci. Technol. Adv. Mater., 2015, 16(4), 044602, DOI:10.1088/1468-6996/16/4/044602.
- E. Mahon, D. R. Hristov and K. A. Dawson, Chem. Commun., 2012, 48, 7970–7972 RSC.
- P. Pietzonka, B. Rothen-Rutishauser, P. Langguth, H. Wunderli-Allenspach, E. Walter and H. P. Merkle, Pharm. Res., 2002, 19, 595–601 CrossRef CAS.
- A. Salvati, C. Aberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818–826 CAS.
- T. Tenuta, M. P. Monopoli, J. Kim, A. Salvati, K. A. Dawson, P. Sandin and I. Lynch, PLoS One, 2011, 6, e25556 CAS.
- I. L. Aye and J. A. Keelan, Chem.-Biol. Interact., 2013, 203, 456–466 CrossRef CAS PubMed.
- H. Ali, I. Kalashnikova, M. A. White, M. Sherman and E. Rytting, Int. J. Pharm., 2013, 454, 149–157 CrossRef CAS PubMed.
- L. Cartwright, M. S. Poulsen, H. M. Nielsen, G. Pojana, L. E. Knudsen, M. Saunders and E. Rytting, Int. J. Nanomed., 2012, 7, 497–510 CAS.
- S. Correia Carreira, L. Walker, K. Paul and M. Saunders, Nanotoxicology, 2013, 9, 66–78 CrossRef PubMed.
- A. R. Menjoge, A. L. Rinderknecht, R. S. Navath, M. Faridnia, C. J. Kim, R. Romero, R. K. Miller and R. M. Kannan, J. Controlled Release, 2011, 150, 326–338 CrossRef CAS PubMed.
- M. S. Poulsen, T. Mose, L. L. Maroun, L. Mathiesen, L. E. Knudsen and E. Rytting, Nanotoxicology, 2013, 9, 79–86 CrossRef PubMed.
- P. Wick, A. Malek, P. Manser, D. Meili, X. Maeder-Althaus, L. Diener, P. A. Diener, A. Zisch, H. F. Krug and U. von Mandach, Environ. Health Perspect., 2010, 118, 432–436 CrossRef CAS PubMed.
- A. I. Ivanov, in Exocytosis and Endocytosis, ed. A. I. Ivanov, Humana Press, Totowa, NJ, 1st edn, 2008, pp. 15–33 Search PubMed.
- L. E. Dike, C. S. Chen, M. Mrksich, J. Tien, G. M. Whitesides and D. E. Ingber, In Vitro Cell. Dev. Biol.: Anim., 1999, 35, 441–448 CrossRef CAS PubMed.
- J. J. Moon, M. S. Hahn, I. Kim, B. A. Nsiah and J. L. West, Tissue Eng., Part A, 2009, 15, 579–585 CrossRef CAS PubMed.
- S. Raghavan, C. M. Nelson, J. D. Baranski, E. Lim and C. S. Chen, Tissue Eng., Part A, 2010, 16, 2255–2263 CrossRef CAS PubMed.
- J. P. Kaiser, T. Buerki-Thurnherr and P. Wick, J. King Saud Univ., Sci., 2013, 25, 15–27 CrossRef.
- A. A. Torrano, J. Blechinger, C. Osseforth, C. Argyo, A. Reller, T. Bein, J. Michaelis and C. Brauchle, Nanomedicine, 2013, 8, 1815–1828 CrossRef CAS PubMed.
- M. Geiser, B. Rothen-Rutishauser, N. Kapp, S. Schürch, W. Kreyling, H. Schulz, M. Semmler, V. I. Hof, J. Heyder and P. Gehr, Environ. Health Perspect., 2005, 113, 1555–1560 CrossRef PubMed.
- F. G. Strobl, D. Breyer, P. Link, A. A. Torrano, C. Brauchle, M. F. Schneider and A. Wixforth, Beilstein J. Nanotechnol., 2015, 6, 414–419 CrossRef PubMed.
- A. A. Torrano and C. Brauchle, Beilstein J. Nanotechnol., 2014, 5, 1616–1624 CrossRef PubMed.
- J. Blechinger, A. T. Bauer, A. A. Torrano, C. Gorzelanny, C. Brauchle and S. W. Schneider, Small, 2013, 9, 3970–3980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Nanoparticle size distribution measurements, fluorescence intensity measurements, cell viability experiments. See DOI: 10.1039/c6ra13782b |
| This journal is © The Royal Society of Chemistry 2016 |