
A first-principles study of the avalanche pressure of alpha zirconium
 *ab
*ab
Abstract
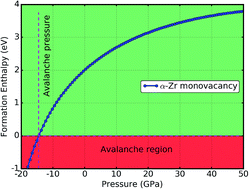
We investigate the stability of a monovacancy in alpha zirconium under various strains and pressures by examining the vacancy formation energy through first-principles calculations. There is a maximum formation energy of 2.35 eV under uniaxial strain corresponding to a c/a ratio of 1.75. Under volumetric strain, the formation energy increases as the strain increases. The formation energy as a function of the volumetric stress or pressure was also examined, with a minimum value of 2.00 eV at zero pressure. Using the equations of state method, we find that the formation volume of the vacancies decreases as the pressure increases, with a value of 0.6 unit-atom-volume at zero pressure. The formation enthalpy increases monotonically as the pressure increases. We predict that the avalanche pressure of alpha zirconium is −15 GPa, where vacancy formation is exothermic, causing avalanche swelling and the failure of the material.
 Please wait while we load your content...
Please wait while we load your content...

